Študija dr. Stephanie Seneff: (str. 26)
Potencial za trajno vključitev gena za beljakovinsko bodico koronavirusa SARS CoV-2 v človeški genom
https://ijvtpr.com/index.php/IJVTPR/article/view/23
*
Povedali so nam, da cepiva SARS-CoV-2 mRNA ni mogoče vključiti v človeški genom, ker se sporočilna RNK ne more spremeniti nazaj v DNK. To je napačno. V človeških celicah obstajajo elementi, imenovani retrotranspozoni LINE-1, ki lahko dejansko integrirajo mRNA v človeški genom z endogeno povratno transkripcijo. Ker je mRNA, ki se uporablja v cepivih, stabilizirana, se dlje časa zadržuje v celicah, kar povečuje možnosti, da se to zgodi. Če je gen za SARS-CoV-2 bodico integriran v del genoma, ki ni utišan in dejansko izraža virusno beljakovino, je mogoče, da bodo ljudje, ki bodo prejeli to cepivo, do konca življenja stalno izražali SARS-CoV-2 bodice iz svojih somatskih celic.
S cepljenjem ljudi s cepivom, ki povzroči, da njihova telesa proizvajajo virusne bodice "in situ", so ljudje dejansko cepljeni s patogenim proteinom. S toksinom, ki lahko povzroči dolgotrajno vnetje, težave s srcem in povečano tveganje za nastanek raka. Dolgoročno lahko povzroči tudi prezgodnjo nevrodegenerativno bolezen.
Vir: https://publishwall.si/narava.zdravi/post/604996/spartacus-dokument-o-bioloskem-orozju-sars-cov-2-nepravilnem-zdravljenju-covid-19-bolnikov-nanotehnologiji-in-transhumanizmu
*
Genetski modificirana RNA molekula iz COVID cjepiva može mijenjati ljudski genom
Tomislav Domazet-Lošo je doktor genetike, izvanredni profesor na Medicinskom fakultetu na Hrvatskom katoličkom sveučilištu i znanstveni suradnik na najvećem hrvatskom znanstveno-istraživačkom centru multidisciplinarna karaktera, Institutu Ruđer Bošković u Zagrebu.
https://dokumentarac.hr/covid-19/video-prof-dr-sc-tomislav-domazet-loso-mrna-cjepiva-zasto-se-zanemaruje-biologija-retropozicije/
*
Kakšna je možnost, da mRNA SARS CoV-2 iz cepiv za COVID-19 spremeni našo DNK?
Avtor: dr. James Weiler Lyons 24. januar 2021
Obstaja verjetnost, da bo vsak, ki bo prejel cepivo na osnovi MRNA proti SARS-CoV-2, doživel spremembe v svoji DNK v celicah, ki jih je "okužila" enkapsulirana mRNA. Obstaja tudi verjetnost, da bodo ljudje, ki prejmejo cepivo proti mRNA, te spremembe prenašali na nekatere ali vse svoje otroke, kar je resnična skrb za nekatere verne ljudi. Živimo v tveganem svetu. Želimo, da nihče ne umre ali zboli "po nepotrebnem" – torej da bi se tveganje za to smrt ali bolezen lahko razumno odpravilo, ali če ne bi bilo odpravljeno, da bi bilo vsaj čim manjše.
Ko je SARS-CoV-2 vstopil v človeško populacijo kot patogen, ga večina iz naše vrste ni videla. Podatki Trevorja Bedforda o starosti SARS CoV-2 in moje raziskave kažejo na dejstvo, da so virusi, podobni SARS-CoV-2, obstajali že leta 2005 ali prej. Če je SARS-CoV-2 vstopil v človeško vrsto pred letom 2019, zagotovo ni učinkoval globalno – morda je povzročil lokalne smrti, povezane s pljučnico, ki se niso pripisale ničemur posebnemu. Pravzaprav se je to v zadnjih dveh ali treh desetletjih morda že večkrat zgodilo. Ker je bil smrtonosen, virus ni dolgo živel. Ko se je leta 2019 zgodil preskok vrste, je bil virus, ki je naredil preskok, bolj prenosljiv in morda malo manj smrtonosen.
Način, kako SARS-CoV-2 ustvarja bolezen pri ljudeh, je zloben. Predvidel sem ne-respiratorne učinke virusa aprila 2020, ko sem objavil svojo študijo, ki uvaja koncept patogenega priminga. Na žalost se zdi, da virus razen na pljuča vpliva tudi na veliko tkiv in organov, vključno z deli našega imunskega sistema in imunskim odzivom na virus, verjetno z razvojem avto-protiteles na beljakovine bodisi v določenih organih, bodisi v nekaterih primerih prisotnih v vseh celicah telesa. Avtoimunost v toliko tkivih se pojavi zaradi ponavljajočih se izpostavljenosti virusnim beljakovinam, kar je pričakovano glede na visoko raven podobnosti virusnih beljakovin z našimi lastnimi beljakovinami – zato sem trdil, da delov virusnih beljakovin ne smemo vključiti v nobeno cepivo.
Še eno vprašanje se mi je porodilo aprila 2020, na katero nisem imel pripravljenega odgovora: Kakšna je verjetnost, da bo ta virus povzročil spremembe na naši DNK? To vprašanje se med znanstveniki zastavi na naslednji način: Ali lahko pričakujemo, da se genom RNK virusa ali kateri koli del virusnega genoma kot DNK kopira v človeški genom?
Dr. Teresa Deisher je strokovnjak za insercijsko mutagenezo – proces, s katerim lahko tuje DNA vstopijo v jedro celice in se vstavijo v človeški ali živalski genom. Aprila 2020 sem jo vprašal, kakšna je verjetnost, da se mRNA, na primer iz cepiva Moderne, prekopira v človeški genom. Njen odgovor je bil: Približno enaka stopnja verjetnosti kot pri virusu.
Če pogledamo razpoložljive študije, najdemo študijo Zhang in sod., (2020), ki jo je objavil Inštitut Whitehead. Študija (5) je objavljena na biorxiv strežniku in do tega pisanja (24.1. 2021) še ni bila strokovno pregledana. Kljub temu je študija pokazala, da se virusni genom lahko reverzno prepiše v DNA, v procesu, ki zahteva poseben encim, imenovan reverzna transkriptaza. Študija kaže, da se lahko virusna DNK (komplementarna virusni RNA) kopira v človeški genom na dva načina: če so človeške celice okužene z virusom HIV, lahko encim reverzne trankriptaze deluje tudi na RNA virusa SARS-CoV-2.
Ugotovili so, da je bil genom SARS-CoV-2 prenešen v nekatere človeške celice, ki niso bile okužene z virusi, ki dajejo beljakovino reverzne transkriptaze. Avtorji te študije so domnevali, da virus aktivira speči encim reverzne transkriptaze v našem genomu, ki je v utišanih starodavnih virusih LINES, ki so se nekoč vključili v genome naših prednikov. (op. prev.: 7% naše DNK so retrovirusi, ki imajo encim reverzne transkriptaze)
Torej se lahko pričakuje, da se virusna RNA vključi v človeški genom. Kaj to pomeni za mRNA cepivo, ki kodira stabilizirano virusno beljakovinsko bodico? Če bodo rezultati študije Whitehead ponovljeni, potem je treba imeti v mislih naslednje: Molekula mRNA, ki kodira stabilizirano beljakovinsko bodico SARS-CoV-2 je molekula RNA – in zato lahko razumno pričakujemo, da jo poberejo kateri koli proteini reverzne transkriptaze.
Ni podatkov, ki bi izključili vključitev mRNA iz cepiv v genom celic pri cepljenih ljudeh, ker to vprašanje ni bilo obravnavano. Prav to vprašanje sta postavila dva ločena udeleženca (dr. Moore in dr. Meissner) na zasedanju Svetovalnega odbora FDA za cepiva in sorodne biološke izdelke, ko so govorili o cepivu Pfizer (ki po Pfizerju vsebuje RNA, ki kodira "celo mutirano beljakovinsko bodico"). Dr. Meissner je posebej vprašal, ali lahko zarodki tvegajo spremembo DNA prek povratne trankriptaze, če bi se bodoče matere cepile. Pfizer in drugi proizvajalci cepiv niso neposredno obravnavali teh vprašanj.
Glede na odkritje študije Zhang in sod., ki je pokazala, da je možno, da virusna RNA spremeni človeški genom, se zdi razumno, da se to lahko zgodi tudi zaradi mRNA iz mRNA cepiv. Kako pogosto se to lahko zgodi, ni znano.
Kaj pa zarodne celice?
Mnogim ljudem bi bilo bolj pomembno vprašanje, ali bi cepivo lahko vodilo do trajnih, dednih sprememb človeškega genoma, z dodajanjem novih variacij, ki se prenašajo iz generacije v generacijo, torej ali cepivo lahko spremeni zarodne celice?
Zarodne celice so spermiji in jajčeca in vsaka sprememba celic, v kateri koli fazi razvoja – vključno, vendar ne samo v fazi zgodnjega razvoja ploda, lahko privede do vnosa nove DNK v človeški genom. Pomembna študija, ki delno obravnava to vprašanje, je študija bio porazdelitve Bahl in sod. (2012), Moderna študija z vprašanjem: Kje v telesu konča enkapsulirana mRNA iz cepiva proti COVID-19? Če tkiva, v katerih tehnologija lokalizira RNA, vključujejo razmnoževalne organe, je mogoče sklepati, da je v nekaterih primerih možna sprememba zarodnih celic. Študija Bahl in sod. iz leta 2012 je uporabljala mRNA, za kodiranje beljakovin iz virusov gripe, vendar je kljub temu dokazala načelo: komercialni produkt mRNA je bil najden v testisih.

Podatkov o lokalizaciji mRNA v jajčnikih ni bilo, niti pri fetusih v brejih miših. Odgovor na vprašanje, ali bi mRNA iz cepiv lahko spremenila linijo človeških zarodnih celic z dednimi variacijami, je "verjetno" – vendar pa spet ni znana pogostost tega dogodka, niti pri kom bi se to lahko zgodilo.
Nevroznanstvenik Chris Shaw je pred kratkim razpravljal o tej študiji z Del Bigtree-jem v oddaji Highwire. Osredotočal se je predvsem na najdbo virusnih beljakovin v možganih in da je verjetno, da povzročijo imunski odziv v možganih tj. potencialno prispevajo k aktivaciji imunskih celic v možganih (mikroglia celic), ki se odzivajo na virusne okužbe ali druge tuje snovi.
Vprašanje, ali bi vas moralo mRNA cepivo skrbeti ali ne, je osebno: če je vaša vera, religija ali osebna želja, da ne bi za vedno spremenili svoje rodbinske genetske linije, bi lahko privolili k drugačni obravnavi okužbe s COVID19, ki okužbo ohranja na minimumu, kot je uporaba zdravil. Če pa verjamete, da je evolucija smiselna s spremembami, ne glede na vir in vas ne moti ideja, da lahko vaši potomci izrazijo COVID19 beljakovinske bodice, potem je okužba ali injekcija lahko vaša priljubljena pot.
Od dr. Anthonyja Faucija pa smo izvedeli, da mRNA cepiva morda ne bodo zagotovila imunitete na SARS-CoV-2 na način, ki preprečuje prenos okužbe. Od dr. Faucija smo izvedeli tudi, da se sedanja cepiva morda ne bodo izkazala za učinkovita proti novim variantam virusa. V teh časih morajo ljudje temeljito iskati v lastnih Ustavah in moralnem kompasu in sami določiti, katere od dveh ali več nezaželenih poti bi imeli raje.
K sreči pa zdaj vemo, da virusna okužba COVID19 lahko daje imuniteto vsaj toliko časa, kot cepivo – kar pomeni, da lahko samovoljna injekcija cepiva po predhodni diagnozi COVID19 služi le za podvojitev tveganja za spremembo genoma in zarodnih celic. Podobno lahko izpostavljenost cepivu povzroči povečano virusno okužbo zaradi cepiva, kot je opisano v članku Ojačanje virusnih okužb zaradi cepiv (Vaccine-induced enhancement of viral infections) (6) v reviji Cepiva iz leta 2009.
Kot evolucijski biolog nisem fatalist. Zame prihodnost prinaša spremembe na podlagi pogojev, v katerih smo se našli, in naših odločitev glede na to, kaj vemo o teh pogojih. V razpravah z ljudmi, ki imajo iskrena verska prepričanja, se zdi, da je pri nekaterih ljudeh, ki se nameravajo razmnoževati, naravni potek, ki pušča rezultate usodi, mogoče ohraniti le z izključitvijo cepiva tj. s preprečevanjem dvojnega tveganja genomskih sprememb. Drugim bo vera v nujne izide pomagala odstraniti strahove in bo njihovo usodo položila v božje roke.
* * *
Ali lahko mRNA cepivo trajno spremeni človeško DNK?
Znanost pravi, da lahko.
Raziskave znanstvenikov na Harvardu in MIT o RNA virusa SARS-CoV-2 kažejo, da bi mRNA cepiva lahko trajno spremenila genomsko DNK, je povedal Doug Corrigan, dr. med. biokemik in molekularni biolog, ki pravi, da je zato potrebno več raziskav.
V običajnih okoliščinah telo naredi ("prepiše") mRNA iz DNK v jedru celice. MRNA nato potuje iz jedra v citoplazmo, kjer daje navodila o tem, katere beljakovine narediti. Cepiva pošljejo svoje kemično sintetizirane mRNA (v paketu z navodili za proizvodnjo beljakovinskih bodic) neposredno v citoplazmo. Po podatkih Centra za nadzor in preprečevanje bolezni (CDC) in večine znanstvenikov se mRNA ustavi v citoplazmi. CDC trdi, da mRNA cepiva "na noben način ne vplivajo oz nimajo interakcije z našo DNK." CDC trdi, da virusna mRNA ne more vstopiti v jedro celice (kjer se nahaja DNK) in da se celica znebi mRNA kmalu po koncu uporabe njenih navodil.
Decembrski predtisk študije o SARS-CoV-2, ki so jo naredili znanstveniki s Harvarda in Massachusetts Institute of Technology (MIT), daje ugotovitve o divjem koronavirusu in delovanju virusne RNA. Študija SARS-CoV-2 RNA reverse-transcribed and integrated into the human genome (8) je nastala na podlagi dejstva, da je veliko število ljudi imelo pozitiven PCR test za COVID-19 še dolgo časa po koncu okužbe. Njihove ključne ugotovitve so bile naslednje: RNA virusa SARS-CoV-2 se lahko reverzibilno prepišejo v človeških celicah, ta DNK zaporedja se lahko vključijo v celični genom in se nato prepišejo (pojav imenovan "retro-integracija") — in obstajajo znane celične poti, ki pojasnjujejo, kako se to zgodi.
Po mnenju doktorja biokemije in molekularne biologije dr. Douga Corrigana, te pomembne ugotovitve (ki so v nasprotju s "trenutno biološko dogmo") spadajo v kategorijo "stvari, ki se absolutno in nedvoumno ne morejo zgoditi, a so se dejansko zgodile."
Ugotovitve raziskovalcev Harvarda in MIT-a so po Besedah Corrigana omajale predpostavke CDC o mRNA cepivih. Mesec dni pred pojavom predprinta študije Harvard-MIT je Corrigan napisal blog, v katerem so opisani možni mehanizmi in poti, po katerih bi lahko mRNA cepiva proizvedla enak pojav. V drugi objavi na blogu, napisani po objavi predprinta študije, je Corrigan poudaril, da imajo ugotovitve Harvard-MIT o koronavirusni RNK velike posledice za mRNA cepiva. Čeprav ne trdi, da se bo sintetična mRNA iz cepiva nujno obnašala enako kot koronavirusna RNK – to je trajno spremenila človeško genomsko DNK – dr. Corrigan meni, da ta možnost obstaja in si zasluži resno obravnavo.
Po Corriganovem mnenju je prispevek predprinta študije v tem, da »potrjuje, da je sprememba DNK verjetna oz. zelo verjetna.«
Reverzna transkripcija
Kot kaže besedna zveza "reverzna transkripcija, pot od DNK do mRNA ni vedno enosmerna. Encimi, imenovani reverzne transkriptaze, lahko pretvorijo RNA v DNK, kar omogoča, da se RNA vključi v DNK v celičnem jedru. Reverzna transkripcija (prepis) ni neobičajna. Genetiki poročajo, da "več kot 40 % genomov sesalcev vsebuje produkte reverzne transkripcije." Preliminarni dokazi, ki so jih navedli raziskovalci Harvard-MIT, kažejo, da lahko endogeni encimi reverzne transkripcije omogočajo povratni prepis koronavirusnih RNA in sprožijo njihovo vključevanje v človeški genom. Avtorji predlagajo, da klinične posledice zahtevajo nadaljnje študije zaradi različnih možnosti, glede na mesto vstavljanja integriranih virusnih fragmentov v človeški genom in zdravstveno stanje posameznika – kar lahko vključuje hujše imunske reakcije ... kot so "citokinska nevihta" ali avtoimunske reakcije."
Študija iz leta 2012 kaže, da ima integracija virusnega genoma v človeško DNK lahko hude posledice za celico, vključno z motnjami genov, insercijsko mutagenezo in smrtjo celice. Corrigan pravi, da možne poti, ki omogočajo povratno (retro) integracijo virusne RNA ali mRNA iz cepiva v človeško DNK, niso neznane ljudem, ki razumejo molekularno biologijo na globljem nivoju."
Kljub temu je razprava v predprintu študije o reverzni transkripciji in integraciji mRNA v človeški genom povzročila negativne komentarje bralcev, ki ne želijo ponovno preučiti biološke dogme, med njimi so nekateri celo zagovarjali umik študije (čeprav so predprinti študij po definiciji neobjavljeni) z razlogom, da bodo »teoretiki zarote« študijo vzeli za "dokaz", da lahko mRNA cepiva dejansko spremenijo našo genetsko kodo. Bolj premišljeni bralci so se strinjali s Corriganom, da študija postavlja pomembna vprašanja. Eden od bralcev je na primer napisal, da manjka potrditvenih dokazov, ki kažejo, da se beljakovinska bodica izraža samo kratek čas (recimo 1-3 dni) po cepljenju," in dodal: "Mislimo, da je tako, vendar za to ni nobenih dokazov."
Pravzaprav je odprto vprašanje, kako dolgo se v celicah ohranjajo sintetične mRNA iz cepiva – in s tem navodila za celice, da ohranijo proizvodnjo virusnih beljakovinskih bodic.
Običajno je RNA krhka in nestabilna molekula. Po mnenju znanstvenikov ta krhkost velja za mRNA katerega koli živega bitja - rastlin, bakterij, virusov ali človeka."
Toda sintetična mRNA v cepivih za COVID-19 je drugačna zgodba. Dejansko je bil korak, ki je znanstvenikom in proizvajalcem cepiv na koncu omogočil, da rešijo svoj desetletja trajajoči neuspeh pri uporabi mRNA, ugotovitev kako kemično spremeniti mRNA, da bi povečali njeno stabilnost in dolgoživost – z drugimi besedami, zdaj proizvajajo RNA, ki v celici traja veliko dlje kot virusna RNA ali celo RNA, ki jo naša celica običajno proizvaja za normalno proizvodnjo beljakovin. Samo ugibamo lahko, kaj sintetična mRNA počne, medtem ko je v celici. Corrigan domneva, da okrepljena dolgoživost sintetične mRNA iz cepiv povečuje verjetnost, da se bo vključila v človeško DNK. Poleg tega, ker je mRNA iz cepiva tudi inženirsko oblikovana tako, da je bolj učinkovita pri prepisu v beljakovine, so lahko negativni učinki mRNA iz cepiva bolj pogosti in močneje izraženi v primerjavi z RNA naravnega virusa.
Corrigan priznava, da lahko nekateri zavrnejo njegova opozorila in rečejo: "Če je virus sposoben to doseči, zakaj bi me potem skrbelo, če cepivo počne isto?" Corrigan ima pripravljen prepričljiv odgovor: »Je velika razlika med scenarijem, kjer se ljudem naključno in nevede spremeni genetika, ker so bili izpostavljeni koronavirusu in med scenarijem, po katerem bodo načrtno cepili milijarde ljudi, medtem ko jim govorijo, da se sprememba njihove genetike ne dogaja.« Na žalost je prevladujoči odnos ta, da "dirka za cepljenje javnosti" upravičuje to dodatno tveganje.
Sredi novembra, potem ko je Jerusalem Post bralcem povedal, da "ko se bo svet začel cepiti s temi povsem novimi in revolucionarnimi cepivi, ne bo vedel praktično nič o njihovih dolgoročnih učinkih," je direktor izraelske bolnišnice trdil, da ni vredno čakati še dve leti, da se ugotovijo "edinstvena in neznana tveganja" mRNA cepiv ali potencialni dolgoročni učinki.
V ZDA je navdušenje nad tehnologijo mRNA podobno. Le nekaj dni po tem, ko je CDC objavil posodobljene podatke, ki kažejo, da je bilo do marca 2021 prijavljenih več kot 2.200 smrtnih primerov posameznikov, ki so prejeli mRNA cepivo Pfizerja ali Moderne, je Atlantic pohvalil to tehnologijo z besedami, da sintetična mRNA iz Pfizer in Moderna COVID-19 cepiv predstavlja dosežek, ki bi lahko spremenil svet. Namesto da bi zavrnili možnost integracije mRNA v človeški genom kot teorijo zarote, bi znanstveniki morali opraviti dodatne študije, da bi ugotovili dejansko tveganje.
Corrigan je na primer prepričan, da »in vitro« podatki v človeških celičnih linijah (eden od virov podatkov, ki so jih preučili raziskovalci Harvard-MIT) ponujajo zanesljive rezultate, vendar je še vedno treba prepričljivo dokazati genomsko spremembo v resničnem življenju s PCR - sekvencioniranjem DNK ali testom Southern Blot prečiščene genomske DNK bolnikov s COVID-19 in cepljenih posameznikov. Toda namesto da bi obravnavala te raziskovalne vrzeli, se korporacije navdušujejo nad potencialom uporabe umetne mRNA, za upravljanje celičnih mehanizmov in proizvodnjo kakršnekoli možne beljakovine.
V sporočilu za javnost 10.marca 2021 so oznanili mRNA cepivo za zmagovalca v boju s COVID-19 in sporočili, da zdaj vsa večja farmacevtska podjetja preizkušajo tehnologijo mRNA in sicer s sprejetjem licenčnih pogodb in/ali sodelovanjem z dobro uveljavljenimi družbami za proizvodnjo RNA. Sodeč po pripravljenosti vodilnih v farmacevtskih družbah, da spregledajo dolgoročna in morda večgeneracijska tveganja zaradi mRNA cepiv, lahko verjamemo, da jih privlačijo vizije povečanja prihodkov z neskončnimi možnostmi poigravanja z mRNA izdelki.
* * *
Prof. dr. sc. Romeo F. Quijano, zaslužni profesor dr. med. je povedal (9), da lahko pride do insercijske mutageneze, kar pomeni naključno vstavljanje virusne mRNA v celični genom človeka, kar lahko sprememni izražanje genov ali aktivira celične onkogene ( možnost indukcije tumorja).
* * *
Imunologinja in molekularna biologinja prof.dr. Dolores Cahill je povedala (10) (18):
»Pogledala sem v EU zakonodajo in videla, da je EU dne 17. julija 2020 sprostila vso zakonodajo na področju gensko spremenjenih organizmov in tako so zdaj vsi testirani in injicirani ljudje gensko spremenjeni organizmi (GSO). To je zato, ker je virusna mRNA v njihovem telesu in ni znano, ali se bo integrirala v gene oz. kromosome in do tega zagotovo prihaja, ker je toliko smrti po cepljenju. Zato potrebujemo shrambo ene na sto vial z mRNA, da vidimo kaj je dejansko v cepivih, ko bodo zlasti starejši ljudje začeli umirati.
Če se mRNA sekvence integrirajo v kromosome, jih bomo izražali do konca življenja. Virusna mRNA gre lako v reproduktivne organe in se prenese na potomce. In če ste človek, ki izraža virusne proteine, ste po zakonski definiciji gensko spremenjen organizem, ki potrebuje dovoljenje za gibanje, tako kot vsak GSO produkt....
Zato je potrebno dobiti zakonit dostop do vial, da se preveri vsebina cepiva in trditve žvižgačev, da so v cepivu še druge vrste mRNA in jaz to zlahka naredim, ker imamo mrežo forenzičnih laboratorijev in smo vsi povezani.«
* * *
Dr.sc. Sanna Ehdin, dr.med., švedska imunologinja, avtorica in lektorica pravi (11):
»Podporniki tega "cepiva" trdijo, da se mRNA razgradi, toda kaj v resnici vedo o učinkih na naš genetski material, na naš imunski sistem in ali bo to spremenilo človekov genetski material? Tega nihče ne ve, ker preprosto ni bilo varnostnih testov ali dolgoročnih študij. Nihče ne prevzema odgovornosti in zagotovo ne farmacevtske industrije. Ta postopek je pravzaprav genetska manipulacija, ki je bila dolgo prepovedana in je doslej veljala za kriminalno. Kako lahko dovolimo, da se to zdaj zgodi?«
* * *
"Genetika oz. specifične genetske analize v tej študiji niso ovrednotene," so priznali v Pfizerjev protokolu. Enako velja za cepivo podjetja Moderna (12)
* * *
Dr. Lee Merritt, ki je preučevala biološka orožja in je delala kot ortopedski kirurg v ameriški mornarici ter je bila članica odbora Medicinske organizacije Arizone ter je objavila številne strokovno preverjene dokumente, meni, da mRNA cepiva proti COVID-19 prerazporejajo naše genetske kode in nas naredijo bolj dojemljive za okužbo z drugimi virusi.(13)
* * *
Prof. Didier Raoult, vodilni mikrobiolog in infektolog (14): Najprej bom predstavil Alana Fischer-ja, ki je en največjih znanstvenikov v Franciji in je izvrsten zdravnik. Za razliko od mnogih, ki le sodelujejo v protokolu testiranja, je on odkril gensko terapijo za imunodeficitne smrtnonosne bolezni otrok. Bil je prvi, ki je to naredil. Vsaka intervencija z nukleinskimi kislinami, DNK in RNK, prinaša nepredvidljive spremembe, kar se je tudi redno dogajalo. dr. Alain Fischer je najbolj merodajna oseba, ki lahko oceni, kako deluje mRNA cepivo.
Na spodaj priloženem videu si lahko pogledate TED Talk iz 2017, v katerem nastopa direktor Moderne – Dr. Tal Zacks. Iz »njegovih ust« lahko slišite, da gre pri uporabi mRNA za nič drugega kot (citat):
“We are actually hacking the software of life.” (dejansko smo ugrabili software življenja)
Ta software je seveda naša DNK in in mRNA je koda, s katero spreminjajo osnovno gradivo človeka.
V nadaljevanju dr. Tal pove tudi:
»V vsaki celici je nekaj, kar se imenuje messenger RNA ali na kratko mRNA, ki prenaša kritične informacije iz DNK v naših genih na beljakovine, ki so v resnici stvari, iz katerih smo vsi narejeni. To so ključne informacije, ki določajo, kaj bo celica naredila. Torej o tem razmišljamo kot o operacijskem sistemu. Torej, če bi to lahko spremenili, če bi lahko uvedli vrstico kode ali spremenili vrstico kode, se izkaže, da ima to globoke posledice za vse, od gripe do raka.«
Če bi lahko uvedli vrstico kode ali spremenili vrstico kode? To je ni čisto nič drugega, kot že omenjeno zgoraj – VDOR v programsko opremo življenja – VDOR v človeški DNK.
* * *
Na prvem delu slike je del navodil, ki ga je Ihan pripravil za NIJZ in so namenjena zdravniški stroki v Sloveniji. Na tej sliki Ihan trdi, da mRNA nikoli ne stopi v jedro celice.
A na drugem delu slike, vzete iz Enciklopedije Britannica, je popolnoma jasno, da se mRNA veže direktno na DNK znotraj jedra celice in da Ihan nesramno laže ter zavaja. Njegova trditev pa se seveda ne sklada tudi s tistim, kar je povedal glavni mož Moderne. Kdo bi vedel, mogoče pa Ihan ve več kot izumitelj sam.
Vir: Denis Hass FB 17.3. 2021
* * *
Belgijski zdravnik dr. Johan Denis: mRNA se lahko vključi v DNK in ga nepovratno spremeni za vse prihodnje generacije (16)
* * *
* * *
Dr. Christian Perrone, profesor za infekcijske in tropske bolezni (17): Normalno gre v celicah sporočilo od DNK do RNK, ampak možno je tudi obratno pri določenih okoliščinah, ker imajo naše celice endogene retroviruse v DNK naših kromosomov, ki lahko proizvedejo encim reverzne transkriptaze. Zato tuja virusna mRNA iz injekcije cepiva lahko kodira DNK in se integrira v naše kromosome.
»Zato obstaja realna nevarnost trajne transformacije naših genov. Obstaja tudi možnost, da se preko modifikacije DNK v jajčecih ali spermijih te genske spremembe prenesejo na potomce.« Prof. Christian Perrone.
* * *
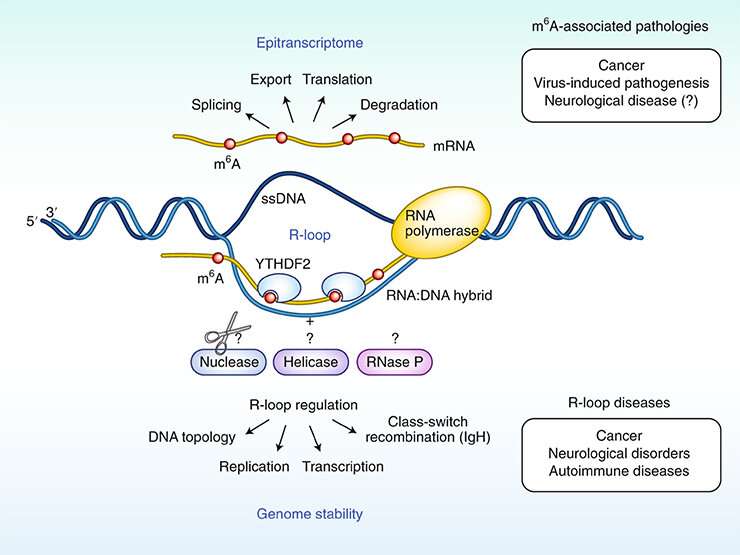
Znanstveni članek (19) z imenom “Modifikacija m6A RNA kot novi igralec pri regulaciji R-zanke“, raziskovalne skupine za dinamično regulacijo genov pod vodstvom Arnea Klunglanda iz IMB-a, objavljen v reviji Nature Genetics.
Po novem sodelovanju med UiO-a in raziskovalnih skupin v Nottinghamu in Oxfordu, se je ugotovilo, da ima RNA neposreden učinek na stabilnost DNK. Mnoge raziskave so pokazale da je regulacija z modifikacijo RNA pomembna pri razvoju raka. Če se odstranijo geni za kemične vezave 6-metiladenina, je rezultat nevro-degeneracija pri miših in ljudeh.
V področjih DNK, kjer se RNA veže za eno od DNK tako, da komplementarna DNK nit postane edina nit (struktura R-zanke), se bo stabilnost DNK spremenila, če je RNA kemično modificirana m6A. Nekaj raziskovalnih skupin sodeluje, da bi preučili kakšen učinek ima to na molekulo DNK. »Znano je že, da so področja R-zanke povezana s sekvencami DNK, ki imajo aktivne gene in da to lahko vodi do preloma kromosomov in izgube genetskih informacij«, je povedal Klungland.
*

*
Običajno se epigenetska regulacija genov preučuje z raziskovanjem dinamičnih modifikacij DNK in proteinov (to so t.i. epigenetske modifikacije). Spremembe lahko vključijo ali izključijo določene gene brez spreminjanja genetske kode. V zadnjih desetih letih so odkrili, da dinamične modifikacije obstajajo tudi v RNA in da imajo pomembno vlogo pri regulaciji genov.
Najbolj pogosta modifikacija mRNA je 6-metiladenin (m6A). Zdaj so ugotovili, da je ta modifikacija pomembna za preživetje celice. Področje raziskav modifikacij RNA se imenuje epitranscriptomics.
Ena prvih študij na tem področju je rezultat sodelovanja med raziskovalci v Chicagu, Pekingu in Oslu (Zheng, Dahl i sur., Molekularna celica, 2012., 49, 18-29).
Vir: https://phys.org/news/2020-01-rna-effect-dna.html
Prihodnost bo pokazala, ali bodo te ugotovitve o vplivu modificirane RNA na spremembo molekule DNK odprle pot za nove raziskave o varnosti neznanih vplivov mRNA cepiv, ki so že v masovni uporabi.
* * *
Študija o vključitvi tuje DNK v človeški genom (20): Illegitimate DNA integration in mammalian cells:
Virusna DNK se lahko integrira v človeški genom z enakim mehanizmom kot vsaka druga tuja DNK, na primer DNK virusa SV40. Genska terapija (op. prev.: kot je mRNA cepivo za COVID-19) lahko povzroči nelegalno integracijo virusnih sekvenc DNK in zato predstavlja tveganje z neznanimi posledicami zaradi genomskih sprememb.
* * *
Prof.dr. Tomislav Terzin je profesor genetike, molekularne biologije in biologije razvoja na Unverzi Alberta v Kanadi. Povedal je (21):
»Pfizer mRNA cepivo je nova tehnologija, ki ima umetno narejeno genetsko informacijo za virusno spike beljakovinsko bodico, kar dajo v lipidne nanodelce. Ali bo ta nanodelec ostal v celični citoplazmi, ali pa bo šel tudi v jedro celice, kjer je DNK- to ni znano.«
* * *
Dr. Michael Yeadon, bivši podpredsednik Pfizerja pravi (22), da ker so gensko terapijo predstavili kot cepivo, niso povedali, kam bo šla mRNA in kako dolgo bo delovala. Kodira beljakovinsko bodico, ki je biološko aktivna in ima tudi fusogensko lastnost zlepljanja več celic, kar lahko povzroči koagulacijo krvi (mikrotromboza).
»Pri vsakem človeku bo mRNA šla v druge dele telesa, običajno tja, kjer je telo ranljivo. Nekateri so bolj občutljivi za tvorbo krvnih strdkov, tudi mlade ženske lahko dobijo vensko trombozo po prejemu mRNA cepiva.«
* * *
Revolucionarni preboj je naredil kanadski znanstvenik Derek Rossi leta 2010 povsem po naključju. Zdaj upokojeni harvardski profesor je v intervjuju za National Post trdil (23), da je našel način, da "reprogramirajo" molekule, ki nosijo genetska navodila za razvoj celic v človeškem telesu in vseh bioloških življenjskih oblikah.
»Te molekule se imenujejo "obveščevalna ribonukleinska kislina" ali mRNA in imajo sposobnost, da na novo napišejo ta navodila za proizvodnjo kakršne koli celice znotraj biološkega organizma. Resnično pomembno odkritje tukaj je bilo, da lahko zdaj uporabite mRNA, in če gre v celice, potem lahko mRNA izrazi katerokoli beljakovino v celicah.«
Malo je verjetno da bo Moderna dobila FDA dovoljenje pred tehnologijo hidrogela, ki ga razvija agencija zvezne vlade DARPA, skupaj z Profusa tehnologijo svetlobnih senzorjev, ki naj bi dobila FDA dovoljenje v prvi polovici leta 2021 in se bo zelo verjetno uporabila v cepivu za COVID-19 z možnostjo spremembe naše DNK.
* * *
Dr. Reid Sheftall,11. marec 2021:Resnica v znanosti: Ne cepite se, dokler ne vidite tega (24):
Strašljivi citati iz strokovno pregledanih študij:
- genomska integracija DNK cepiv v kromosome gostitelja je še ena skrb glede biološke varnosti, saj to lahko vodi v mutagenezo (mutacije DNK) in onkogenezo (rakotvornost)
- čeprav so prejšnje študije pokazale, da je tveganje insercije vsebine cepiva v kromosome gostitelja precej nizko, ameriška Uprava za hrano in zdravila (FDA) in Svetovna zdravstvena organizacija (WHO) priporočata, da so študije o integraciji del varnostnega programa DNA cepiv
- v virusnih vektorskih cepivih so rekombinantni virusi, ki kodirajo antigene v nepovezanem gensko spremenjenem virusu. Astra Zeneca uporablja človeški adenovirus, ki se ne replicira, Johnson&Johnson pa adenovirus primatov (šimpanzov). Za koronaviruse se najbolj pogosto uporablja Encefalitis virus kopitarjev.
- rekombinantni virusi nosijo tveganje integracije svojega genoma v človeški genom, zato so pred pričetkom kliničnih preizkusov potrebne dodatne ocene biološke varnosti cepiv
Če encim reverzne transkriptaze spremeni sintetično mRNA iz cepiva v DNK, se ta lahko integrira v genom cepljenje osebe. Rekombinantni virus je virus, v katerega vstavijo del mRNA za kodiranje beljakovinske bodice virusa SARS CoV-2. To je podobno kot pri mRNA cepivih
Viri:
(9) https://www.bulatlat.com/2020/08/21/hazards-of-the-covid-19-vaccine/
(10) https://www.youtube.com/watch?v=vca1uVerXGQ
(15) https://www.youtube.com/watch?v=FU-cqTNQhMM&t=7s
(16) https://www.facebook.com/KraljevicMarkoIII/videos/2918869551724897
(18) https://www.facebook.com/101017414921399/videos/425883341954748
(19) https://dokumentarac.hr/novosti/modificirana-mrna-ima-izravan-ucinak-na-gene/
(20) https://www.nature.com/articles/3302074
(24) https://www.youtube.com/watch?v=2xUxCIfMjIQ
*
Dr. Sherri Tennpeny: mRNA iz cepiv se lahko vključi v človeški genom (8.00 min):
https://www.brighteon.com/d7f6e042-e3e0-4e42-b5be-d40a0c4bac73
*
Cepivo mPNA Pfizer-BionTech spreminja genetski profil, ki uravnava ekspresijo citokinov ...
Cepivo mRNA BNT162b2 proti SARS-CoV-2 reprogramira tako prilagodljive kot prirojene imunske odzive
Cepivo mRNA BNT162b2 (Pfizer / BionTech) proti SARS-CoV-2 reprogramira tako prilagojene (specifične) kot prirojene imunske odzive.
V tej vnaprej natisnjeni študiji (ki jo je treba še strokovno pregledati) obeti niso preveč pomirjujoči za tiste, ki so medicino že od nekdaj jemali resno.Z drugimi besedami, cepiva mRNA spremenijo genetski profil, ki uravnava izražanje citokinov . Zelo resna stvar je to, kljub dejstvu, da sta varnost in učinkovitost teh cepiv zelo vprašljivo ocenjeni na približno 95%.
"... Cepivo proti mRNA Pfizer / BioNTech BNT162b2 je bilo prvo registrirano cepivo COVID-19 in se je izkazalo za do 95% učinkovito (na podlagi katerih meril, ki potrjujejo to" učinkovitost ", ni znano) pri preprečevanju SARS- Okužbe s CoV-2. O splošnih učinkih nove vrste cepiv mRNA je malo znanega, zlasti če imajo kombinirane učinke na prirojene in pridobljene (specifične) imunske odzive . Tu smo potrdili, da je cepljenje zdravih posameznikov z BNT162b2 povzročilo humoralno in celično imunost, učinkovito proti več različicam SARS-CoV-2.
Zanimivo pa je, da je cepivo BNT162b2 moduliralo tudi proizvodnjo vnetnih citokinov v prirojenih imunskih celicah tako na specifične (SARS-CoV-2) kot nespecifične (virusne, glivične in bakterijske) dražljaje.
Odziv prirojenih imunskih celic na ligande TLR4 in TLR7 / 8 je bil po cepljenju z BNT162b2 manjši, medtem ko so bili glivično povzročeni citokinski odzivi močnejši.
*
Skratka, cepivo mRNA BNT162b2 povzroči kompleksno funkcionalno reprogramiranje prirojenih imunskih odzivov, kar je treba upoštevati pri razvoju in uporabi tega novega razreda cepiv ... "
https://vk.com/@davide_suraci-il-vaccino-pfizer-biontech-a-mrna-modifica-il-profilo-geneti
https://www.medrxiv.org/content/10.1101/2021.05.03.21256520v1
The BNT162b2 mRNA vaccine against SARS-CoV-2 reprograms both adaptive and innate immune responses
Summary
The mRNA-based BNT162b2 vaccine from Pfizer/BioNTech was the first registered COVID-19 vaccine and has been shown to be up to 95% effective in preventing SARS-CoV-2 infections. Little is known about the broad effects of the new class of mRNA vaccines, especially whether they have combined effects on innate and adaptive immune responses. Here we confirmed that BNT162b2 vaccination of healthy individuals induced effective humoral and cellular immunity against several SARS-CoV-2 variants. Interestingly, however, the BNT162b2 vaccine also modulated the production of inflammatory cytokines by innate immune cells upon stimulation with both specific (SARS-CoV-2) and non-specific (viral, fungal and bacterial) stimuli. The response of innate immune cells to TLR4 and TLR7/8 ligands was lower after BNT162b2 vaccination, while fungi-induced cytokine responses were stronger. In conclusion, the mRNA BNT162b2 vaccine induces complex functional reprogramming of innate immune responses, which should be considered in the development and use of this new class of vaccines.
*
*
Zakaj se magneti lepijo na mesto vboda injekcije s COVID-19 cepivom?
https://odysee.com/@TimTruth:b/Magneticcovidvaxarm-1:6?src=open
*
mRNA cepivo povzroči smrt telesnih celic (apoptosis) : https://odysee.com/@shortXXvids:e/Dr-VS-K-why-m-RNA-vacine-different:2?src=open&r=BWSeHJ8asYoo4uRLsWhCb8D17qQ2xW22&fbclid=IwAR0KJ1_GHe6ea7VDbXafSbcMk1eajvL6g22jnV6he7Ggnyl3FvOHY20savE
*
Dr. Christian Velot: vrste Covid-19 cepiv (min 21.00): Cepivo Astra Zenece in Sputnik uporablja vektor adenovirus z rekombinantno DNK (ima del DNK adenovirusa in DNK SARS CoV-2 za kodiranje beljakovinske bodice). Ker je to DNK cepivo, obstaja nevarnost, da se virusna DNK vključi v naše kromosome. (min 30.00). Imamo že slabe izkušnje z rekombinantnimi virusi pri uporabi genske terapije. V študiji genske terapije na desetih otrocih, sta dva imela resne težave, ker se je DNK iz zdravila vstavila na napačno mesto v kromosomu, ki vsebuje onkogene (rakave gene). Tako sta dva otroka zaradi genske terapije dobila levkemijo. O tem so poročali leta 2003. Tudi takrat so uporabili rekombinantni adenovirus, kot v cepivih za COVID-19. To se imenuje insercijska mutageneza, kar pomeni da se povzroči mutacija genov z vstavljanjem tuje DNK. Bile so težave tudi pri imunoterapiji za zdravljenje raka, kjer se skuša aktivirati imunski odziv, za prepoznavanje in uničenje rakavih celic. Tudi ta terapija uporablja rekombinantni adenovirus. V več kliničnih raziskavah v Belgiji je takšna imunoterapija povzročila imunotoksičnost = neželjen imunski odziv, kot je avtoimunska bolezen ali kritični sistemski vnetni odziv, zaradi katerega je umrla ena oseba med 18, ki so sodelovale pri testiranju.
Pri mRNA in pri vektorskih DNA cepivih za COVID-19 obstaja nevarnost virusne rekombinacije. Virusi iste vrste med seboj izmenjujejo genetski material in lahko se zgodi, da postanejo bolj virulentni. Tak virus je H1N1, ki je kombinacija ptičje, prašičje in človeške gripe. To se zgodi, ko je celica okužena z dvema vrstama virusov. Pri genetskih cepivih se lahko zgodi, da bo v celici genski material iz cepiva in nekega drugega virusa. Možnost, da se to zgodi je sicer zelo majhna, pri eni osebi na 10 ali 100 milijonov. Vendar če bo cepljenih več sto ali milijarde ljudi, se bo to verjetno zgodilo in lahko se pojavi nov, močnejši virus. Tako se je začela tudi pandemija SARS CoV-2.
https://www.youtube.com/watch?v=YZM2Gekxy2I
*
https://www.pnas.org/content/118/21/e2105968118
Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues
See all authors and affiliations
-
Contributed by Rudolf Jaenisch, April 19, 2021 (sent for review March 29, 2021; reviewed by Anton Berns and Anna Marie Skalka)
Significance
An unresolved issue of SARS-CoV-2 disease is that patients often remain positive for viral RNA as detected by PCR many weeks after the initial infection in the absence of evidence for viral replication. We show here that SARS-CoV-2 RNA can be reverse-transcribed and integrated into the genome of the infected cell and be expressed as chimeric transcripts fusing viral with cellular sequences. Importantly, such chimeric transcripts are detected in patient-derived tissues. Our data suggest that, in some patient tissues, the majority of all viral transcripts are derived from integrated sequences. Our data provide an insight into the consequence of SARS-CoV-2 infections that may help to explain why patients can continue to produce viral RNA after recovery.
Abstract
Prolonged detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA and recurrence of PCR-positive tests have been widely reported in patients after recovery from COVID-19, but some of these patients do not appear to shed infectious virus. We investigated the possibility that SARS-CoV-2 RNAs can be reverse-transcribed and integrated into the DNA of human cells in culture and that transcription of the integrated sequences might account for some of the positive PCR tests seen in patients. In support of this hypothesis, we found that DNA copies of SARS-CoV-2 sequences can be integrated into the genome of infected human cells. We found target site duplications flanking the viral sequences and consensus LINE1 endonuclease recognition sequences at the integration sites, consistent with a LINE1 retrotransposon-mediated, target-primed reverse transcription and retroposition mechanism. We also found, in some patient-derived tissues, evidence suggesting that a large fraction of the viral sequences is transcribed from integrated DNA copies of viral sequences, generating viral–host chimeric transcripts. The integration and transcription of viral sequences may thus contribute to the detection of viral RNA by PCR in patients after infection and clinical recovery. Because we have detected only subgenomic sequences derived mainly from the 3′ end of the viral genome integrated into the DNA of the host cell, infectious virus cannot be produced from the integrated subgenomic SARS-CoV-2 sequences.
Continuous or recurrent positive severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) PCR tests have been reported in samples taken from patients weeks or months after recovery from an initial infection (1⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓⇓–17). Although bona fide reinfection with SARS-CoV-2 after recovery has recently been reported (18), cohort-based studies with subjects held in strict quarantine after they recovered from COVID-19 suggested that at least some “re-positive” cases were not caused by reinfection (19, 20). Furthermore, no replication-competent virus was isolated or spread from these PCR-positive patients (1⇓–3, 5, 6, 12, 16), and the cause for the prolonged and recurrent production of viral RNA remains unknown. SARS-CoV-2 is a positive-stranded RNA virus. Like other beta-coronaviruses (SARS-CoV-1 and Middle East respiratory syndrome-related coronavirus), SARS-CoV-2 employs an RNA-dependent RNA polymerase to replicate its genomic RNA and transcribe subgenomic RNAs (21⇓⇓–24). One possible explanation for the continued detection of SARS-CoV-2 viral RNA in the absence of virus reproduction is that, in some cases, DNA copies of viral subgenomic RNAs may integrate into the DNA of the host cell by a reverse transcription mechanism. Transcription of the integrated DNA copies could be responsible for positive PCR tests long after the initial infection was cleared. Indeed, nonretroviral RNA virus sequences have been detected in the genomes of many vertebrate species (25, 26), with several integrations exhibiting signals consistent with the integration of DNA copies of viral mRNAs into the germline via ancient long interspersed nuclear element (LINE) retrotransposons (reviewed in ref. 27). Furthermore, nonretroviral RNA viruses such as vesicular stomatitis virus or lymphocytic choriomeningitis virus (LCMV) can be reverse transcribed into DNA copies by an endogenous reverse transcriptase (RT), and DNA copies of the viral sequences have been shown to integrate into the DNA of host cells (28⇓–30). In addition, cellular RNAs, for example the human APP transcripts, have been shown to be reverse-transcribed by endogenous RT in neurons with the resultant APP fragments integrated into the genome and expressed (31). Human LINE1 elements (∼17% of the human genome), a type of autonomous retrotransposons, which are able to retro-transpose themselves and other nonautonomous elements such as Alu, are a source of cellular endogenous RT (32⇓–34). Endogenous LINE1 elements have been shown to be expressed in aged human tissues (35) and LINE1-mediated somatic retrotransposition is common in cancer patients (36, 37). Moreover, expression of endogenous LINE1 and other retrotransposons in host cells is commonly up-regulated upon viral infection, including SARS-CoV-2 infection (38⇓–40).
In this study, we show that SARS-CoV-2 sequences can integrate into the host cell genome by a LINE1-mediated retroposition mechanism. We provide evidence that the integrated viral sequences can be transcribed and that, in some patient samples, the majority of viral transcripts appear to be derived from integrated viral sequences.
Results
Integration of SARS-CoV-2 Sequences into the DNA of Host Cells in Culture.
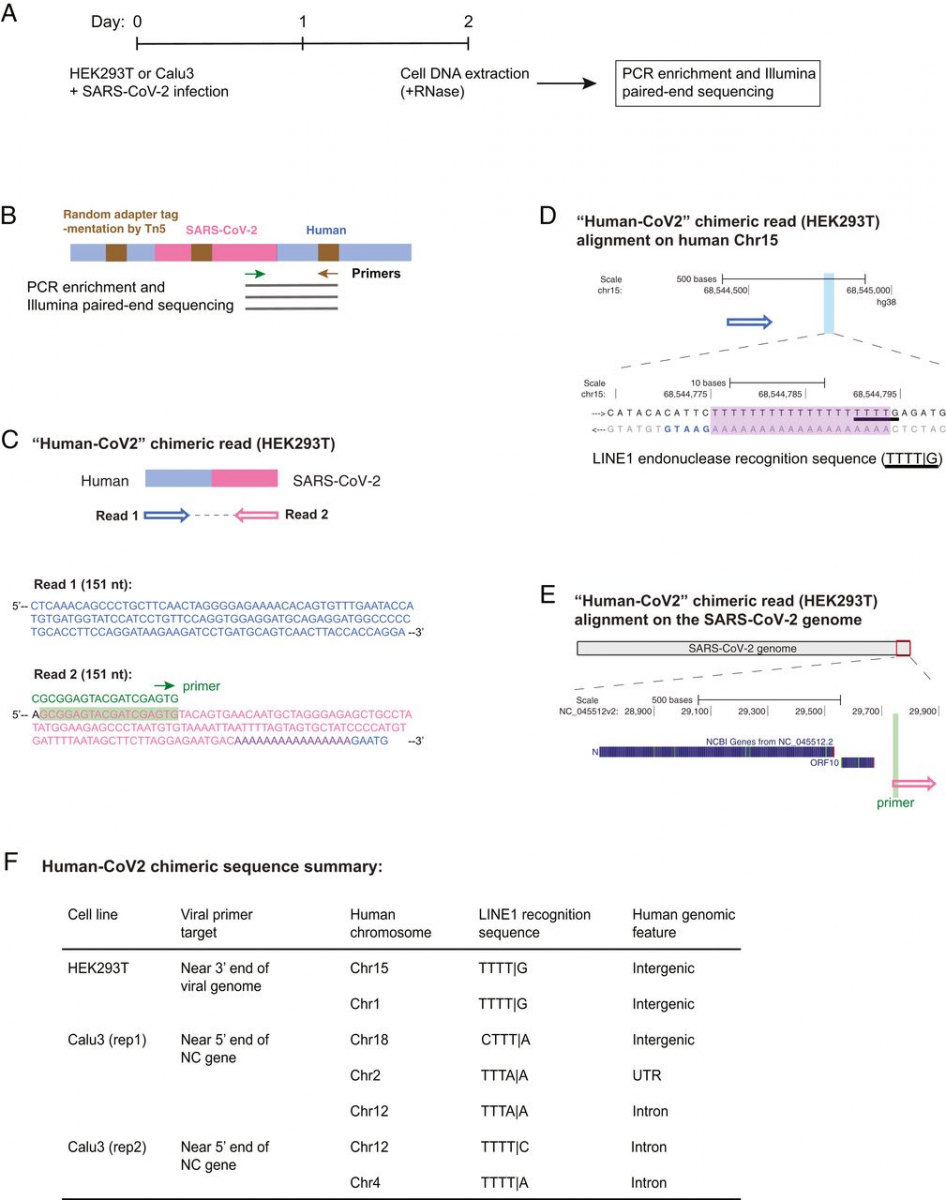
We used three different approaches to detect genomic SARS-CoV-2 sequences integrated into the genome of infected cells. These approaches were Nanopore long-read sequencing, Illumina paired-end whole genomic sequencing, and Tn5 tagmentation-based DNA integration site enrichment sequencing. All three methods provided evidence that SARS-CoV-2 sequences can be integrated into the genome of the host cell.
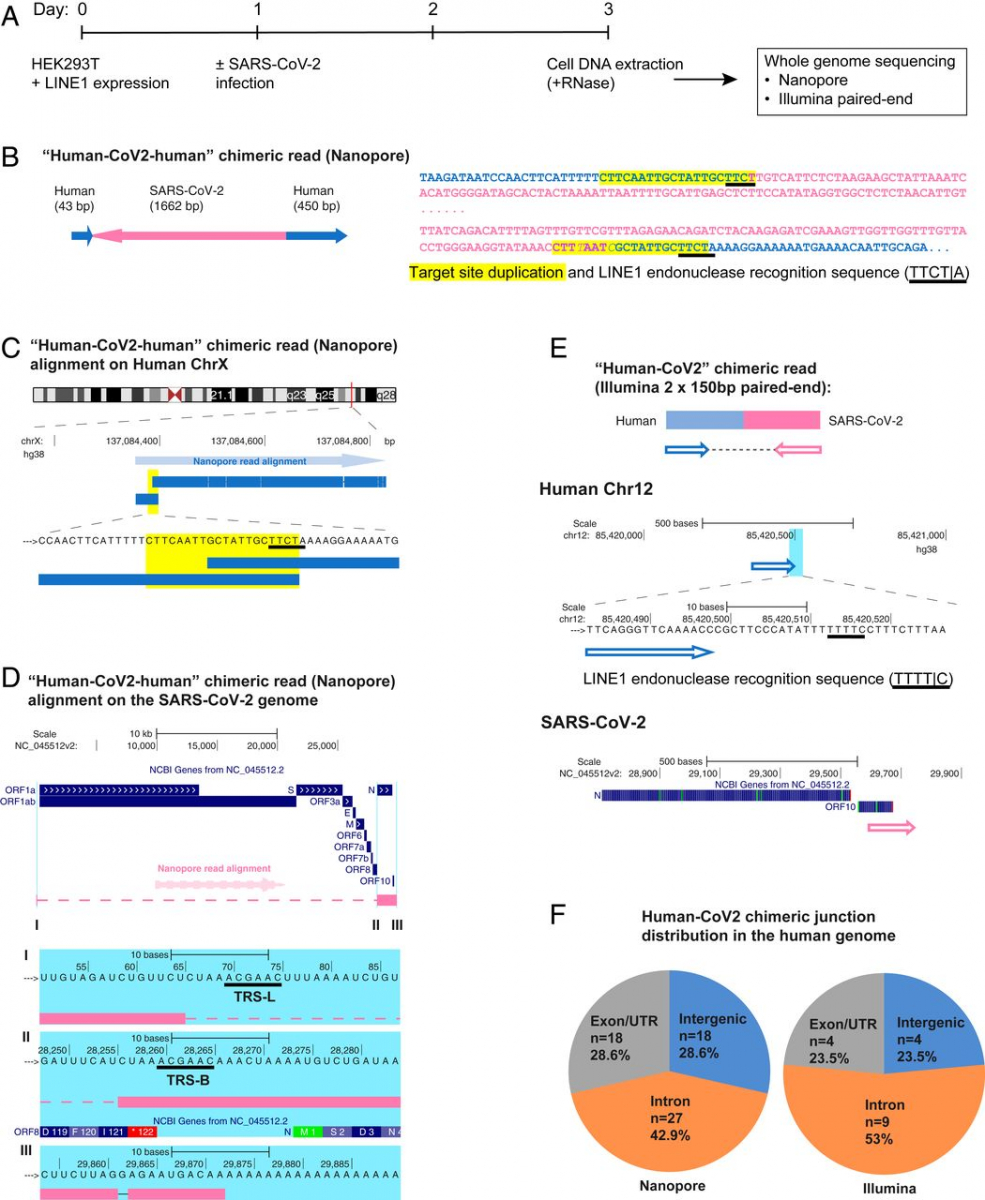
To increase the likelihood of detecting rare integration events, we transfected HEK293T cells with LINE1 expression plasmids prior to infection with SARS-CoV-2 and isolated DNA from the cells 2 d after infection (SI Appendix, Fig. S1A). We detected DNA copies of SARS-CoV-2 nucleocapsid (NC) sequences in the infected cells by PCR (SI Appendix, Fig. S1B) and cloned the complete NC gene (SI Appendix, Fig. S1D) from large-fragment cell genomic DNA that had been gel-purified (SI Appendix, Fig. S1C). The viral DNA sequence (NC) was confirmed by Sanger sequencing (Dataset S1). These results suggest that SARS-CoV-2 RNA can be reverse-transcribed, and the resulting DNA could be integrated into the genome of the host cell.
To demonstrate directly that the SARS-CoV-2 sequences were integrated into the host cell genome, DNA isolated from infected LINE1-overexpressing HEK293T cells was used for Nanopore long-read sequencing (Fig. 1A). Fig. 1 B–D shows an example of a full-length viral NC subgenomic RNA sequence (1,662 bp) integrated into the cell chromosome X and flanked on both sides by host DNA sequences. Importantly, the flanking sequences included a 20-bp direct repeat. This target site duplication is a signature of LINE1-mediated retro-integration (41, 42). Another viral integrant comprising a partial NC subgenomic RNA sequence that was flanked by a duplicated host cell DNA target sequence is shown in SI Appendix, Fig. S2 A–C. In both cases, the flanking sequences contained a consensus recognition sequence of the LINE1 endonuclease (43). These results indicate that SARS-CoV-2 sequences can be integrated into the genomes of cultured human cells by a LINE1-mediated retroposition mechanism. Table 1 summarizes all of the linked SARS-CoV-2–host sequences that were recovered. DNA copies of portions of the viral genome were found in almost all human chromosomes. In addition to the two examples given in Fig. 1 and SI Appendix, Fig. S2, we also recovered cellular sequences for 61 integrants for which only one of the two host–viral junctions was retrieved (SI Appendix, Fig. S2 D–F and Table 1; Nanopore reads containing the chimeric sequences summarized in Dataset S2). Importantly, about 67% of the flanking human sequences included either a consensus or a variant LINE1 endonuclease recognition sequence (such as TTTT/A) (SI Appendix, Fig. S2 D–F and Table 1). These LINE1 recognition sequences were either at the chimeric junctions that were directly linked to the 3′ end (poly-A tail) of viral sequences, or within a distance of 8–27 bp from the junctions that were linked to the 5′ end of viral sequences, which is within the potential target site duplication. Both results are consistent with a model in which LINE1-mediated retroposition provides a mechanism to integrate DNA copies of SARS-CoV-2 subgenomic fragments into host genomic DNA. About 71% of the viral sequences were flanked by intron or intergenic cellular sequences and 29% by exons (Fig. 1F and Table 1). Thus, the association of the viral sequences with exons is much higher than would be expected for random integration into the genome [human genome: 1.1% exons, 24% introns, and 75% intergenic DNA (44)], suggestive of preferential integration into exon-associated target sites. While previous studies showed no preference for LINE1 retroposition into exons (45, 46), our finding suggests that LINE1-mediated retroposition of some other RNAs may be different. We noted that viral–cellular boundaries were frequently close to the 5′ or 3′ untranslated regions (UTRs) of the cellular genes, suggesting that there is a preference for integration close to promoters or poly(A) sites in our experimental system.

Fig. 1.
SARS-CoV-2 RNA can be reverse transcribed and integrated into the host cell genome. (A) Experimental workflow. (B) Chimeric sequence from a Nanopore sequencing read showing integration of a full-length SARS-CoV-2 NC subgenomic RNA sequence (magenta) and human genomic sequences (blue) flanking both sides of the integrated viral sequence. Features indicative of LINE1-mediated “target-primed reverse transcription” include the target site duplication (yellow highlight) and the LINE1 endonuclease recognition sequence (underlined). Sequences that could be mapped to both genomes are shown in purple with mismatches to the human genomic sequences in italics. The arrows indicate sequence orientation with regard to the human and SARS-CoV-2 genomes as shown in C and D. (C) Alignment of the Nanopore read in B with the human genome (chromosome X) showing the integration site. The human sequences at the junction region show the target site, which was duplicated when the SARS-CoV-2 cDNA was integrated (yellow highlight) and the LINE1 endonuclease recognition sequence (underlined). (D) Alignment of the Nanopore read in B with the SARS-CoV-2 genome showing the integrated viral DNA is a copy of the full-length NC subgenomic RNA. The light blue highlighted regions are enlarged to show TRS-L (I) and TRS-B (II) sequences (underlined, these are the sequences where the viral polymerase jumps to generate the subgenomic RNA) and the end of the viral sequence at the poly(A) tail (III). These viral sequence features (I–III) show that a DNA copy of the full-length NC subgenomic RNA was retro-integrated. (E) A human–viral chimeric read pair from Illumina paired-end whole-genome sequencing. The read pair is shown with alignment to the human (blue) and SARS-CoV-2 (magenta) genomes. The arrows indicate the read orientations relative to the human and SARS-CoV-2 genomes. The highlighted (light blue) region of the human read mapping is enlarged to show the LINE1 recognition sequence (underlined). (F) Distributions of human–CoV2 chimeric junctions from Nanopore (Left) and Illumina (Right) sequencing with regard to features of the human genome.
To confirm the integration of SARS-CoV-2 sequences into genomic DNA by another method, we subjected DNA isolated from LINE1-transfected and SARS-CoV-2–infected HEK293T cells to Illumina paired-end whole-genome sequencing, using a Tn5-based library construction method (Illumina Nextera) to avoid ligation artifacts. Viral DNA reads were concentrated at the 3′ end of the SARS-CoV-2 genome (SI Appendix, Fig. S3). We recovered 17 viral integrants (sum of two replicates), by mapping human–viral chimeric DNA sequences (Fig. 1E and Table 2, chimeric sequences summarized in Dataset S3); 7 (41%) of the junctions contained either a consensus or a variant LINE1 recognition sequence in the cellular sequences near the junction (Fig. 1E and Table 2), consistent with a LINE1-mediated retroposition mechanism. Similar to the results obtained from Nanopore sequencing, about 76% of the viral sequences were flanked by intron or intergenic cellular sequences and 24% by exons (Fig. 1F and Table 2).
About 32% of SARS-CoV-2 sequences (6/21 integration events in Nanopore, 4/10 in Illumina data) were integrated at LINEs, short interspersed nuclear elements, or long terminal repeat elements without evidence for a LINE1 recognition site, suggesting that there may be an alternative reverse transcription/integration mechanism, possibly similar to that reported for cells acutely infected with LCMV, which resulted in integrated LCMV sequences fused to intracisternal A-type particle (IAP) sequences (29).
To assess whether genomic integration of SARS-CoV-2 sequences could also occur in infected cells that did not overexpress RT, we isolated DNA from virus-infected HEK293T and Calu3 cells that were not transfected with an RT expression plasmid (Fig. 2A). Tn5 tagmentation-mediated DNA integration site enrichment sequencing (47, 48) (Fig. 2B and SI Appendix, Fig. S4A) detected a total of seven SARS-CoV-2 sequences fused to cellular sequences in these cells (sum of three independent infections of two cell lines), all of which showed LINE1 recognition sequences close to the human–SARS-CoV-2 sequence junctions (Fig. 2 C–F and SI Appendix, Fig. S4 B–D, chimeric sequences summarized in Dataset S4).

Fig. 2.
Evidence for integration of SARS-CoV-2 cDNA in cultured cells that do not overexpress a reverse transcriptase. (A) Experimental workflow. (B) Experimental design for the Tn5 tagmentation-mediated enrichment sequencing method used to map integration sites in the host cell genome. (C) A human–viral chimeric read pair supporting viral integration. The reads are aligned with the human (blue) and SARS-CoV-2 (magenta) genomic sequences. The arrows indicate the read orientations relative to the human and SARS-CoV-2 genomes as shown in D and E. Sequence of the viral primer used for enrichment is shown with green highlight in the read (corresponding to the green arrow illustrated in B). Sequences that could be mapped to both genomes are shown in purple. (D) Alignment of the read pair in C with the human genome (chromosome 15, blue arrow). The highlighted (light blue) region of the human sequence is enlarged to show the LINE1 recognition sequence (underlined) with a 19-base poly-dT sequence (purple highlight) that could be annealed by the viral poly-A tail for “target-primed reverse transcription.” Additional 5-bp human sequence (GAATG, blue) was captured in read 2 (C), supporting a bona fide integration site. (E) Alignment of the read pair in C with the SARS-CoV-2 genome (magenta). The viral primer sequence is shown with green highlight. (F) Summary of seven human–viral chimeric sequences identified by the enrichment sequencing method in the two cell lines showing the integrated human chromosomes, LINE1 recognition sequences close to the chimeric junction, and human genomic features at the read junction.
Expression of Viral–Cellular Chimeric Transcripts in Infected Cultured Cells and Patient-Derived Tissues.
To investigate the possibility that SARS-CoV-2 sequences integrated into the genome can be expressed, we analyzed published RNA-seq data from SARS-CoV-2–infected cells for evidence of chimeric transcripts (49). Examination of these datasets (50⇓⇓⇓⇓–55) (SI Appendix, Fig. S5) revealed a number of human–viral chimeric reads (SI Appendix, Fig. S6 A and B). These occurred in multiple sample types, including cultured cells and organoids from lung/heart/brain/stomach tissues (SI Appendix, Fig. S6B). The abundance of the chimeric reads positively correlated with viral RNA level across the sample types (SI Appendix, Fig. S6B). Chimeric reads generally accounted for 0.004–0.14% of the total SARS-CoV-2 reads in the samples. A majority of the chimeric junctions mapped to the sequence of the SARS-CoV-2 NC gene (SI Appendix, Fig. S6 C and D). This is consistent with the finding that NC RNA is the most abundant SARS-CoV-2 subgenomic RNA (56), making it the most likely target for reverse transcription and integration. However, recent data showed that up to 1% of RNA-seq reads from SARS-CoV-2–infected cells can be artifactually chimeric as a result of RT switching between RNA templates, which can occur during the cDNA synthesis step in the preparation of a RNA-seq library (57). Thus, because there is a mixture of host mRNAs and positive-strand viral mRNAs in infected cells, the identification of genuine chimeric viral–cellular RNA transcripts is compromised by the generation of artifactual chimeras in the assays.
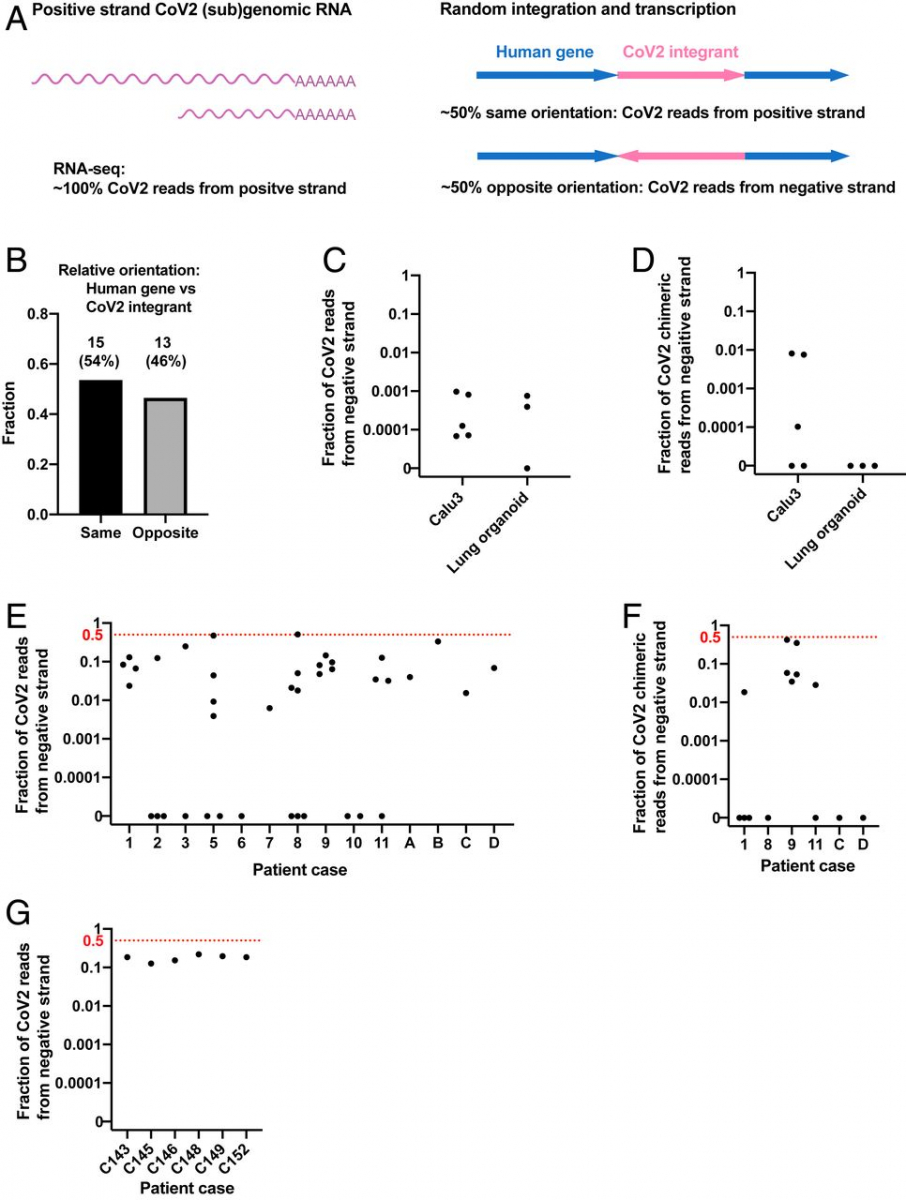
We reasoned that the orientation of an integrated DNA copy of SARS-CoV-2 RNA should be random with respect to the orientation of the targeted host gene, predicting that about half the viral DNAs that were integrated into an expressed host gene should be in an orientation opposite to the direction of the host cell gene’s transcription (Fig. 3A). As predicted, ∼50% of viral integrants in human genes were in the opposite orientation relative to the host gene in our Nanopore dataset (integration at human genes with LINE1 recognition sequences, Fig. 3B). Thus, for chimeric transcripts derived from integrated viral sequences, we would expect that ∼50% of the chimeric transcripts should contain negative-strand viral sequences linked to positive-strand host RNA sequences. We therefore determined the fraction of the viral and human–viral chimeric transcripts in infected cultured cells/organoids and in patient-derived tissues containing negative-strand viral RNA sequences.
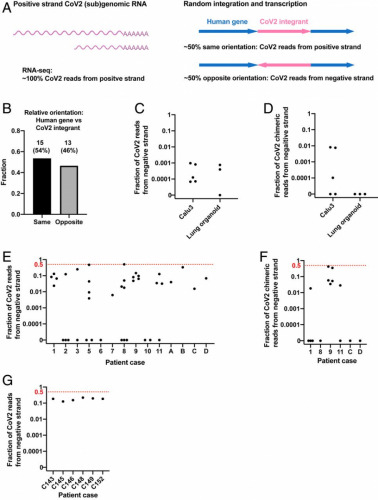
Negative-strand viral RNA-seq reads suggest that integrated SARS-CoV-2 sequences are expressed. (A) Schema predicting fractions of positive- or negative-strand SARS-CoV-2 RNA-seq reads that are derived from viral (sub)genomic RNAs or from transcripts of integrated viral sequences. The arrows (Right) showing the orientation of an integrated SARS-CoV-2 (magenta) positive strand relative to the orientation of the host cellular gene (blue). (B) Fractions of SARS-CoV-2 sequences integrated into human genes with same (n = 15) or opposite (n = 13) orientation of the viral positive strand relative to the positive strand of the human gene. A total of 28 integration events at human genes with LINE1 endonuclease recognition sequences were identified from our Nanopore DNA sequencing of infected LINE1-overexpressing HEK293T cells (Fig. 1A). (C) Fraction of total viral reads that are derived from negative-strand viral RNA in acutely infected cells or organoids (see SI Appendix, Table S1 for details). (D) Fraction of human–viral chimeric reads that contain viral sequences derived from negative-strand viral RNA in acutely infected cells or organoids (see SI Appendix, Table S1 for details). (E) Fraction of total viral reads that are derived from negative-strand viral RNA in published patient RNA-seq data (autopsy FFPE samples, GSE150316, samples with no viral reads or of low library strandedness quality not included; see SI Appendix, Table S2 for details; reanalysis results consistent with the original publication). (F) Fraction of human–viral chimeric reads that contain viral sequences derived from negative-strand viral RNA in published patient RNA-seq data (autopsy FFPE samples, GSE150316; see SI Appendix, Table S2 for details). (G) Fraction of total viral reads that are derived from negative-strand viral RNA in published patient RNA-seq data (BALF samples, GSE145926; see SI Appendix, Table S3 for details). The red dashed lines in E–G indicate the level at which 50% of all viral reads (E and G) or viral sequences in human–viral chimeric reads (F) were from negative-strand viral RNAs, a level expected if all the viral sequences were derived from integrated sequences.
The replication of SARS-CoV2 RNA requires the synthesis of negative-strand viral RNA, which serves as template for replication of viral genomic RNA and transcription of viral subgenomic positive-strand RNA (21). To assess the prevalence of negative-strand viral RNA in acutely infected cells, we determined the ratio of total positive to negative-strand RNAs. Between 0 and 0.1% of total viral reads were derived from negative-strand RNA in acutely infected Calu3 cells or lung organoids [our data and published data (50, 58)] (Fig. 3C and SI Appendix, Table S1), similar to what has been reported in clinical samples taken early after infection (59). These results argue that the level of negative-strand viral RNA is at least 1,000-fold lower than that of positive-strand viral RNA in acutely infected cells, due at least in part to a massive production of positive-strand subgenomic RNA during viral replication. This greatly reduces the likelihood that random template switching during the reverse transcription step in the RNA-seq library construction would generate a large fraction of the artifactual chimeric reads that would contain viral negative-strand RNA fused to cellular positive-strand RNA sequences. We determined that between 0 and 1% of human–viral chimeric reads contained negative-strand viral sequences in the acutely infected cells/organoids (Fig. 3D and SI Appendix, Table S1), consistent with a small fraction of viral reads being derived from integrated SARS-CoV-2 sequences.
In contrast to the results obtained with acutely infected Calu3 cells or lung organoids, up to 51% of all viral reads, and up to 42.5% of human–viral chimeric reads, were derived from the negative-strand SARS-CoV-2 RNA in some patient-derived tissues [published data (60, 61), patient clinical background available in the original publications] (Fig. 3 E–G and SI Appendix, Tables S2 and S3). Single-cell analysis of patient lung bronchoalveolar lavage fluid (BALF) cells from patients with severe COVID [published data (61)] showed that up to 40% of all viral reads were derived from the negative-strand SARS-CoV-2 RNA (SI Appendix, Fig. S7). Fractions of negative-strand RNA in tissues from some patients were orders of magnitude higher than those in acutely infected cells or organoids (Fig. 3 C–G). In fixed (formalin-fixed, paraffin-embedded [FFPE]) autopsy samples, in 4 out of 14 patients (Fig. 3E and SI Appendix, Table S2), and in BALF samples, in 4 out of 6 patients (Fig. 3G and SI Appendix, Table S3), at least ∼20% of the viral reads were derived from negative-strand viral RNA. In contrast to acutely infected cells (Fig. 3 C and D and SI Appendix, Table S1), there was little or no evidence for virus reproduction in these autopsy samples (60). As summarized in SI Appendix, Table S2, there were negative-strand viral sequences in a large fraction of the human–viral chimeric reads (up to ∼40%) in samples from one patient. Different samples derived from the same patient revealed a similarly high fraction of negative viral strand–human RNA reads. Several other patient samples revealed lower fraction of negative viral strand RNA–human RNA chimeras, which were, however, still significantly higher than what was found in acutely infected cells (Fig. 3 D and F and SI Appendix, Table S1 and S2). Because the ability to identify viral–human chimeric reads using short-read RNA-seq is limited, our analysis failed to show significant numbers of chimeric reads in patient BALF samples (SI Appendix, Table S3). In summary, our data suggest that in some patient-derived tissues, where the total number of SARS-CoV-2 sequence-positive cells may be small, a large fraction of the viral transcripts could have been transcribed from SARS-CoV-2 sequences integrated into the host genome.
Discussion
We present here evidence that SARS-CoV-2 sequences can be reverse-transcribed and integrated into the DNA of infected human cells in culture. For two of the integrants, we recovered “human–viral–human” chimeric reads encompassing a direct target site repeat (20 or 13 bp), and a consensus recognition site of the LINE1 endonuclease was present on both ends of the host DNA that flanked the viral sequences. These and other data are consistent with a target primed reverse transcription and retroposition integration mechanism (41, 42) and suggest that endogenous LINE1 RT can be involved in the reverse transcription and integration of SARS-CoV-2 sequences in the genomes of infected cells.
Approximately 30% of viral integrants analyzed in cultured cells lacked a recognizable nearby LINE1 endonuclease recognition site. Thus, it is also possible that integration can occur by another mechanism. Indeed, there is evidence that chimeric cDNAs can be produced in cells acutely infected with LCMV by copy choice with endogenous IAP elements during reverse transcription. This mechanism is expected to create a chimeric cDNA complementary to both LCMV and IAP. In some cases, the resulting chimeric cDNAs were integrated without the generation of a target site duplication (29). A recent study has also suggested that the interaction between coronavirus sequences and endogenous retrotransposon could be a potential viral integration mechanism (40).
It will be important, in follow-up studies, to demonstrate the presence of SARS-CoV-2 sequences integrated into the host genome in patient tissues. However, this will be technically challenging because only a small fraction of cells in any patient tissues are expected to be positive for viral sequences (61). Consistent with this notion, it has been estimated that only between 1 in 1,000 and 1 in 100,000 mouse cells infected with LCMV either in culture or in the animal carried viral DNA copies integrated into the genome (30). In addition, only a fraction of patients may carry SARS-CoV-2 sequences integrated in the DNA of some cells. However, with more than 140 million humans infected with SARS-CoV-2 worldwide (as of April, 2021), even a rare event could be of significant clinical relevance. It is also challenging to estimate the frequency of retro-integration events in cell culture assays since infected cells usually die and are lost before sample collection. For the same reason, no clonal expansion of integrated cells is expected in acute infection experiments. Moreover, the chance of integration at the same genomic locus in different patients/tissues may be low, due to a random integration process.
The presence of chimeric virus–host RNAs in cells cannot alone be taken as strong evidence for transcription of integrated viral sequences because template switching can happen during the reverse transcription step of cDNA library preparation. However, we found that only a very small fraction (0–1%) of chimeric reads from acutely infected cells contained negative-strand viral RNA sequences, whereas, in the RNA-seq libraries prepared from some patients, the fraction of total viral reads, and the fraction of human–viral chimeric reads that were derived from negative-strand SARS-CoV-2 RNAs was substantially higher. For retrotransposon-mediated integration events, the orientation of the reverse-transcribed SARS-CoV-2 RNA should be random with respect to the orientation of a host gene. Thus, for chimeric RNAs derived from integrated viral sequences, about half of the chimeric reads will link positive-strand host RNA sequences to negative-strand viral sequences. In some patient samples, negative-strand viral reads accounted for 40–50% of the total viral RNA sequences and a similar fraction of the chimeric reads contained negative-strand viral RNA sequences, suggesting that the majority if not all of the viral RNAs in these samples were derived from integrated viral sequences.
It is important to note that, because we have detected only subgenomic sequences derived mainly from the 3′ end of the viral genome integrated into the DNA of the host cell, infectious virus cannot be produced from such integrated subgenomic SARS-CoV-2 sequences. The possibility that SARS-CoV-2 sequences can be integrated into the human genome and expressed in the form of chimeric RNAs raises several questions for future studies. Do integrated SARS-CoV-2 sequences express viral antigens in patients and might these influence the clinical course of the disease? The available clinical evidence suggests that, at most, only a small fraction of the cells in patient tissues express viral proteins at a level that is detectable by immunohistochemistry. However, if a cell with an integrated and expressed SARS-CoV-2 sequences survives and presents a viral- or neo-antigen after the infection is cleared, this might engender continuous stimulation of immunity without producing infectious virus and could trigger a protective response or conditions such as autoimmunity as has been observed in some patients (62, 63). The presence of LCMV sequences integrated in the genomes of acutely infected cells in mice led the authors to speculate that expression of such sequences “potentially represents a naturally produced form of DNA vaccine” (30). It is not known how many antigen-presenting cells are needed to elicit an antigen response, but derepressed LINE1 expression, induced by viral infection or by exposure to cytokines (38⇓–40), may stimulate SARS-CoV-2 integration into the genome of infected cells in patients. More generally, our results suggest that integration of viral DNA in somatic cells may represent a consequence of a natural infection that could play a role in the effects of other common disease-causing RNA viruses such as dengue, Zika, or influenza virus.
Our results may also be relevant for current clinical trials of antiviral therapies (64). If integration and expression of viral RNA are fairly common, reliance on extremely sensitive PCR tests to determine the effect of treatments on viral replication and viral load may not always reflect the ability of the treatment to fully suppress viral replication because the PCR assays may detect viral transcripts that derive from viral DNA sequences that have been stably integrated into the genome rather than infectious virus.
Materials and Methods
Cell Culture and Plasmid Transfection.
HEK293T cells were obtained from ATCC (CRL-3216) and cultured in DMEM supplemented with 10% heat-inactivated FBS (HyClone; SH30396.03) and 2 mM L-glutamine (MP Biomedicals; IC10180683) following ATCC’s method. Calu3 cells were obtained from ATCC (HTB-55) and cultured in EMEM (ATCC; 30-2003) supplemented with 10% heat-inactivated FBS (HyClone; SH30396.03) following ATCC’s method.
Plasmids for human LINE1 expression, pBS-L1PA1-CH-mneo (CMV-LINE-1), was a gift from Astrid Roy-Engel, Tulane University Health Sciences Center, New Orleans, LA (Addgene plasmid #51288 ; http://addgene.org/51288; RRID:Addgene_51288) (65); EF06R (5′UTR-LINE-1) was a gift from Eline Luning Prak, University of Pennsylvania, Philadelphia, PA (Addgene plasmid #42940 ; http://addgene.org/42940; RRID:Addgene_42940) (66). Transfection was done with Lipofectamine 3000 (Invitrogen; L3000001) following manufacturer’s protocol.
SARS-CoV-2 Infection.
SARS-CoV-2 USA-WA1/2020 (GenBank: MN985325.1) was obtained from BEI Resources and expanded and tittered on Vero cells. Cells were infected in DMEM plus 2% FBS for 48 h using a multiplicity of infection (MOI) of 0.5 for infection of HEK293T cells and an MOI of 1 or 2 for Calu3 cells. All sample processing and harvest with infectious virus were done in the BSL3 facility at the Ragon Institute.
Nucleic Acids Extraction and PCR Assay.
Cellular DNA extraction was done using a published method (31). For purification of genomic DNA, total cellular DNA was fractionated on a 0.4% (wt/vol) agarose/1× TAE gel for 1.5 h with a 3 V/cm voltage, with λ DNA-HindIII Digest (NEB; N3012S) as size markers. Large fragments (>23.13 kb) were cut out, frozen in −80 °C, and then crushed with a pipette tip. Three volumes (vol/wt) of high T-E buffer (10 mM Tris–10 mM EDTA, pH 8.0) were added, and then NaCl was added to give a final concentration of 200 mM. The gel solution was heated at 70 °C for 15 min with constant mixing and then extracted with phenol:chloroform:isoamyl alcohol (25:24:1, vol/vol/vol) (Life Technologies; 15593031) and chloroform:isoamyl alcohol 24:1 (Sigma; C0549-1PT). DNA was precipitated by the addition of sodium acetate and isopropyl alcohol. For samples with low DNA concentration, glycogen (Life Technologies; 10814010) was added as a carrier to aid precipitation.
RNA extraction was done with RNeasy Plus Micro Kit (Qiagen; 74034) following manufacturer’s protocol.
To detect DNA copies of SARS-CoV-2 sequences, we chose four NC gene-targeting PCR primer sets that are used in COVID-19 tests [SI Appendix, Fig. S1A, primer source from World Health Organization (67), modified to match the genome version of NC_045512.2]. See SI Appendix, Table S4 for PCR primer sequences used in this study. PCR was done using AccuPrime Taq DNA Polymerase, high fidelity (Life Technologies; 12346094). PCR products were run on 1% or 2% (wt/vol) agarose gel to show amplifications.
Nanopore DNA Sequencing and Analysis.
A total of 1.6 μg of DNA extracted from HEK293T cells transfected with the pBS-L1PA1-CH-mneo (CMV-LINE-1) plasmid and infected with SARS-CoV-2 was used to make a sequencing library with the SQK-LSK109 kit (Oxford Nanopore Technologies) and sequenced on one R9 PromethION flowcell (FLO-PRO002) for 3 d and 5 min. The sequencing data were base-called using Guppy 4.0.11 (Oxford Nanopore Technologies) using the high-accuracy model.
Nanopore reads were mapped using minimap2 (68) (version 2.15) with parameters “-p 0.3 -ax map-ont” and a fasta file containing the human genome sequence from ENSEMBL release 93 (ftp://ftp.ensembl.org/pub/release-93/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz) concatenated to the SARS-CoV-2 sequence, GenBank ID: MN988713.1, “Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/IL-CDC-IL1/2020, complete genome.” From the SAM file, we selected all the sequences that mapped to the viral genome and divided them into groups based on the human chromosomes they mapped to. We blasted the selected sequences, using blastn, against a BLAST database made with the human and virus sequences described above. We parsed the blast output into a text file containing one row per high-scoring segment pair (HSP) with a custom perl script. We further filtered that file, for each sequence, by selecting all the viral HSPs and the top three human HSPs. We inspected those files visually to identify sequences containing human–viral–human or human–viral junctions. For a few sequences, longer than 30 kb, we inspected the top 15 human HSPs. Additionally, we visually inspected all the identified reads containing human and viral sequences by the University of California, Santa Cruz (UCSC) BLAT (69) tool. Due to errors in Nanopore sequencing and/or base-calling, artifactual “hybrid sequences” exist in a subset of these reads, sometimes with Watson and Crick strands from the same DNA fragment present in the same read. Therefore, we only focused on chimeric sequences showing clear human–viral junctions and analyzed known LINE1-mediated retroposition features such as target-site duplications and LINE1 endonuclease recognition sequences for evidence of integration.
Tn5 Tagmentation-Mediated Integration Site Enrichment.
We used a tagmentation-based method to enrich for viral integration sites (47, 48). Briefly, we used Tn5 transposase (Diagenode; C01070010) to randomly tagment the cellular DNA with adapters (adapter A, the Illumina Nextera system). Tagmentation was done using 100 ng of DNA for 10 min at 55 °C, followed by stripping off the Tn5 transposase from the DNA with SDS. We used a reverse primer targeting the near-5′ end of SARS-CoV-2 NC gene (CCAAGACGCAGTATTATTGGGTAAA) or a forward primer targeting the near-3′ end of SARS-CoV-2 genome (CTTGTGCAGAATGAATTCTCGTAACT) to linearly amplify (PCR0, 45 cycles) the tagmented DNA fragments containing viral sequences. We took the product of PCR0 and amplified the DNA fragments containing adapter and viral sequences (potential integration sites) using 15–20 cycles of PCR1, with a barcoded (i5) Nextera primer (AATGATACGGCGACCACCGAGATCTACACNNNNNNNNTCGTCGGCAGCGTC, NNNNNNNN indicates the barcode) against the adapter sequence and a viral primer. The viral primer was designed to either target the near-5′ end of SARS-CoV-2 NC gene (GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGCCGACGTTGTTTTGATCG, viral sequence underlined) or target the near-3′ end of SRAS-CoV-2 genome (GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCGCGGAGTACGATCGAGTG, viral sequence underlined). The viral primer also contained an adapter sequence for further PCR amplification. We amplified the PCR1 product by 15–20 cycles of PCR2, using a short primer (AATGATACGGCGACCACCGA) against the i5 Nextera primer sequence and a barcoded (i7) Nextera primer (CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTCTCGTGGGCTCGG, NNNNNNNN indicates the barcode) against the adapter sequence introduced by the viral primer in PCR1. The final product of the PCR2 amplification was fractionated on 1.5% agarose gel (Sage Science; HTC1510) with PippinHT (Sage Science; HTP0001) and 500- to 1,000-bp pieces were selected for Illumina paired-end sequencing. All three PCR steps (PCR0–PCR2) were done with KAPA HiFi HotStart ReadyMix (KAPA;KK2602).
Illumina DNA Sequencing and Analysis.
We constructed libraries for HEK293T cell whole-genome sequencing using the Tn5-based Illumina DNA Prep kit (Illumina; 20018704). The whole-genome sequencing libraries or the libraries from Tn5-mediated integration site enrichment after sizing (described above) were subjected to Illumina sequencing. qPCR was used to measure the concentrations of each library using KAPA qPCR library quant kit according to the manufacturer’s protocol. Libraries were then pooled at equimolar concentrations, for each lane, based on qPCR concentrations. The pooled libraries were denatured using the Illumina protocol. The denatured libraries were loaded onto an SP flowcell on an Illumina NovaSeq 6000 and run for 2 × 150 cycles according to the manufacturer’s instructions. Fastq files were generated and demultiplexed with the bcl2fastq Conversion Software (Illumina).
To identify human–SARS-CoV-2 chimeric DNA reads, raw sequencing reads were aligned with STAR (70) (version 2.7.1a) to a human plus SARS-CoV-2 genome made with a fasta file containing the human genome sequence version hg38 with no alternative chromosomes concatenated to the SARS-CoV-2 sequence from National Center for Biotechnology Information (NCBI) reference sequence NC_045512.2. The following STAR parameters were used to call chimeric reads: –alignIntronMax 1 \–chimOutType Junctions SeparateSAMold WithinBAM HardClip \–chimScoreJunctionNonGTAG 0 \–alignSJstitchMismatchNmax -1–1 -1–1 \–chimSegmentMin 25 \–chimJunctionOverhangMin 25 \–outSAMtype BAM SortedByCoordinate. We extracted viral reads from the generated BAM file by samtools (71) (version 1.11) using command: samtools view -b Aligned.sortedByCoord.out.bam NC_045512v2 > NC_Aligned.sortedByCoord.out.bam. We extracted human–viral chimeric reads by using the read names from the STAR generated Chimeric.out.junction file to get the read alignments from the STAR generated Chimeric.out.sam file by Picard (http://broadinstitute.github.io/picard), using command: java -jar picard.jar FilterSamReads I = Chimeric.out.sam O = hv-Chimeric.out.sam READ_LIST_FILE = hv-Chimeric.out.junction.ids FILTER = includeReadList. We further confirmed each of the chimeric reads and filtered out any unconvincing reads (too short or aligned to multiple sites of the human genome) by visual inspection with the UCSC BLAT (69) tool. We also loaded the STAR generated Aligned.sortedByCoord.out.bam file or the NC_Aligned.sortedByCoord.out.bam file containing extracted viral reads to the UCSC browser SARS-CoV-2 genome (NC_045512.2) to search for additional chimeric reads that were missed by the STAR chimeric calling method. To generate genome coverage file, we used the bamCoverage from the deepTools suite (72) (version 3.5.0) to convert the STAR generated Aligned.sortedByCoord.out.bam file to a bigwig file binned at 10 bp, using command: bamCoverage -b Aligned.sortedByCoord.out.bam -o Aligned.sortedByCoord.out.bw–binSize 10.
RNA-Seq and Analysis.
To identify human–SARS-CoV-2 chimeric reads, published RNA-seq data were downloaded from Gene Expression Omnibus (GEO) with the accession numbers GSE147507 (50), GSE153277 (51), GSE156754 (52), GSE157852 (53), GSE153684 (54), and GSE154998 (55) (summarized in SI Appendix, Fig. S5C). Raw sequencing reads were aligned with STAR (70) (version 2.7.1a) to human plus SARS-CoV-2 genome and transcriptome made with a fasta file containing the human genome sequence version hg38 with no alternative chromosomes concatenated to the SARS-CoV-2 sequence from NCBI reference sequence NC_045512.2, and a gtf file containing the human gene annotations from ENSEMBL version GRCh38.97 concatenated to the SARS-CoV-2 gene annotations from NCBI (http://hgdownload.soe.ucsc.edu/goldenPath/wuhCor1/bigZips/genes/). The following STAR parameters (56) were used to call chimeric reads unless otherwise specified (SI Appendix, Fig. S5C):–chimOutType Junctions SeparateSAMold WithinBAM HardClip \–chimScoreJunctionNonGTAG 0 \–alignSJstitchMismatchNmax -1–1 -1–1 \–chimSegmentMin 50 \–chimJunctionOverhangMin 50.
For RNA-seq strandedness analysis, we generated RNA-seq data using RNA from SARS-CoV-2–infected Calu3 cells. Stranded libraries were constructed with the Kapa mRNA HyperPrep kit (Roche; 08098115702). Libraries were qPCR'ed using a KAPA qPCR library quant kit as per manufacturer’s protocol. Libraries were then pooled at equimolar concentrations, for each lane, based on qPCR concentrations. The pooled libraries were denatured using the Illumina protocol. The denatured libraries were loaded onto an HiSeq 2500 (Illumina) and sequenced for 120 cycles from one end of the fragments. Basecalls were performed using Illumina offline basecaller (OLB) and then demultiplexed. We downloaded published RNA-seq data (stranded libraries) from GEO with the accession numbers GSE147507 (50) (Calu3, SI Appendix, Table S1), GSE148697 (58) (lung organoids, SI Appendix, Table S1), and GSE150316 (60) (patient FFPE tissues, SI Appendix, Table S2). Raw RNA-seq reads were aligned as described above, using parameters–chimSegmentMin 30 \–chimJunctionOverhangMin 30 to call chimeric reads. We extracted total viral reads and human–viral chimeric reads as described above. We convert the viral read BAM files into Bed files using the bamToBed utility in BEDTools (73). We then counted the total and stranded read numbers in the converted BED files.
Published single-cell RNA-seq data were downloaded from GEO with the accession number GSE145926 (61) (patient BALF samples, SI Appendix, Table S3). For bulk analysis, duplicate reads with the same read1 (UMI) and read2 sequences in raw fastq files were removed by dedup_hash (https://github.com/mvdbeek/dedup_hash). Then the pool of read2 were aligned as described above, using parameters –chimSegmentMin 30 \–chimJunctionOverhangMin 30 to call chimeric reads. Read strandedness was analyzed as described above. For single-cell analysis, we generated a custom genome by Cell Ranger (10× Genomics Cell Ranger 3.0.2) (74) mkref, using a fasta file containing the human genome sequence from ENSEMBL release 93 (ftp://ftp.ensembl.org/pub/release-93/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz) concatenated to the SARS-CoV-2 sequence, GenBank ID: MN988713.1, and a gtf file containing human and viral annotations. Read mapping, assigning reads to cell barcodes and removing PCR duplicates were done with Cell Ranger (10× Genomics Cell Ranger 4.0.0) (74) count, using the custom genome described above. We processed the counts using Seurat (version 3.2.2) (75). We removed cells that had less than 200 genes detected or more than 20% of transcript counts deriving from the mitochondria. For each cell, we counted the number of reads mapping to either the positive or negative viral strand.
Data Availability
All data supporting the findings of this study are available within the article and supporting information. All sequencing data generated in this study have been deposited to the Sequence Read Archive, https://www.ncbi.nlm.nih.gov/sra (accession no. PRJNA721333). All published data analyzed in this study are cited in this article with accession methods provided in Materials and Methods.
Acknowledgments
We thank members in the laboratories of R.J. and R.A.Y. and other colleagues from Whitehead Institute and Massachusetts Institute of Technology (MIT) for helpful discussions and resources. We thank Thomas Volkert and staff from the Whitehead genomics core, and Stuart Levine from the MIT/Koch Institute BioMicro center for sequencing support. We thank Lorenzo Bombardelli for sharing protocol and advice for Tn5 tagmentation-mediated integration enrichment sequencing. We thank Jerold Chun, Inder Verma, Joseph Ecker, and Daniel W. Bellott for discussion and suggestions. This work was supported by grants from the NIH to R.J. (1U19AI131135-01; 5R01MH104610-21) and by a generous gift from Dewpoint Therapeutics and from Jim Stone. S.H.H. was supported by the Intramural Research Program of the Center for Cancer Research of the National Cancer Institute. Finally, we thank Nathans Island for inspiration.
Footnotes
- ↵1To whom correspondence may be addressed. Email: jaenisch@wi.mit.edu.
*
SARS-CoV-2 RNA reverse-transcribed and integrated into the human genome
https://pubmed.ncbi.nlm.nih.gov/33330870/
*
Članica Odbora za etiko v Parizu in strokovnjakinja za cepiva dr. Alexandra Henrion Caude o neetičnosti vektorskih gensko spremenjenih cepiv (Astra Zeneca, Janssen, Sputnik) in spreminjanju človeške genetike z mRNA cepivi (Pfizer, Moderna): od 46.00 min dalje: https://www.youtube.com/watch?v=UNjaRWfwz1Q
*
Cepivi AstraZenece in Johnson & Johnsona z adenovirusnim vektorjem gensko zaporedje virusnih proteinov S pošiljajo v celično jedro namesto v citoplazmo, kjer virus običajno proizvaja proteine, so ugotovili nemški znanstveniki v študiji, objavljeni v sredo na povezavi https://www.researchsquare.com/article/rs-558954/v1.
Ko so proteini v jedru, razpadejo, pri čemer nastanejo mutirane različice virusnega proteina S. Ti pa se ne morejo pripeti na celično membrano, kjer poteka imunizacija. Celice jih pošljejo v telo, kar sproži redke motnje strjevanja krvi, so zapisali v študiji, ki je drugi strokovnjaki še niso pregledali.
https://www.researchsquare.com/article/rs-558954/v1
*
Dr. Stephanie Seneff, raziskovalka iz MIT: retrovirusi (tudi koronavirus SARS CoV-2 je retrovirus) se lahko vpišejo v človeško DNK. Tudi sama celica lahko vključi RNA v svojo DNK.
https://www.audible.com/pd/Dr-Stephanie-Seneff-COVID-19-Vaccines-Potential-Effects-on-Human-DNA-Podcast/B093FXWCLC
*
Niso znane posledice genske terapije mRNA za COVID-19, pravi prof. dr. sc. Hadži-Tanovič: Gost emisije Jutro, jutarnjeg programa Prve srpske televizije je prof.dr.sc. Višeslav Hadži-Tanović, dr.med.spec.kardiolog, koji je specijalizirao kardiologiju na Klinici Lariboisier u Parizu kod poznatog kardiologa Buvrena, zatim u Hustonu, na Texas Heart Institute-u i u Švedskoj na Klinici Karolinska Hospital! Redovni je profesor kardiologije Američkog Medicinskog fakulteta (US Medical School). Akademik, aktivan član akademije znanosti u New Yorku. Akademik, član Srpske kraljevske akademije inovatora i znanstvenika. Član je Teksaškog insituta za srce u Huston-u, te član europskog udruženja kardiologa.
*
Človeške celice lahko pretvorijo RNA v DNA . Študijo, objavljeno v Science Advance, ki tvega, da bo treba prepisovati knjige o biologiji, ker ruši dogmo o molekularni biologiji, je pripravila skupina, ki jo vodi Guruhankar Chandramouly z ameriške univerze Thomas Jefferson . In lahko pomaga v boju proti raku.
«To je odkritje, ki ima, če bo potrjeno - genetik Edoardo Boncinelli komentira Ansa - velik vpliv tako na teoretični kot na praktični ravni. Pravzaprav smo že od petdesetih let prejšnjega stoletja vedeli, da se biološke informacije v človeških celicah prenašajo iz DNK v RNK in nato dosežejo proizvodnjo beljakovin. Tako pomemben in neizpodbiten mehanizem, da velja za "dogmo biologije". V zadnjih letih se je ta gotovost začela nihati, zdaj pa se zdi, da je treba v tej študiji poudariti, da bodo potrebne nadaljnje preiskave, ki bodo popolnoma porušene. "
Tumorji so odkrili stikalo za izklop replikacije celic
S preučevanjem posebne različice polimeraze, molekularnih strojev, ki se na splošno ukvarjajo s popravilom verige DNA , so raziskovalci ugotovili, da so tudi theta polimeraze sposobne pretvarjati sekvence Rna .znotraj DNK z mehanizmom, podobnim mehanizmu, ki ga HIV uporablja za spreminjanje naših celic. Po mnenju raziskovalcev se zdi, da se RNA uporablja kot "referenčna knjiga" za zmanjšanje napak, ki so lahko prisotne v verigi DNA. "Odkritje, ki dokazuje, kako veliko je še tisto, česar še ne vemo, in da bi lahko imelo veliko posledic za razvoj genskega inženiringa," je dodal Boncinelli. "To novo odkritje - poudaril je Stefano Gustincich, znanstveni direktor nekodirajočih RNA in RNA na Italijanskem tehnološkem inštitutu (Iit) - ima lahko na medicinskem področju izreden pomen, ker so mehanizmi polimeraze neposredno povezani s širjenjem raka celic in njihova odpornost na zdravljenje.
*
- Sean O Cleirign je ovbjavio zaista bomba vijest.
Nekoliko istaknutih liječnika, stručnjaka za zdravstvo i dobrobit Sons of Liberty Media, Kate Shemirani, njezin kolega dr. Kevin Corbett i japretpostavljali smo da bi trenutna eksperimentalna injekcija mRNA za koronavirus, zvana COV-19, mogla promijeniti nečiji genetski kod ili DNA. Bill Gates je to izjavio, što je bilo uključeno u mom videu“Human Genome 8 and mRNA Vaccine” na Brighteon.com
To je jedan od razloga što se pojam “eksperimentalno ubrizgavanje mRNA s humanim genomom” koristi za opisivanje uboda koji je stavljen u većini javnosti. Iako su mnogi u medijima dr. Anthony Fauci i njegova vesela skupina kroničnih lažova i “provjerivači činjenica” ovu tvrdnju proglasili lažnom, videozapis razgovora TEDx Beacon Street,Tal Zaksa, izvršnog direktora medicinske službea Moderne, Inc. , jedna farmaceutska tvrtka koja proizvodi eksperimentalnu injekciju mRNA tehnologije, potvrđuje da injekcija mRNA za COV-19 može promijeniti vaš genetski kod ili DNA.
https://theirishsentinel.com/2021/03/21/bombshell-moderna-chief-medical-officer-admits-mrna-alters-dna/
U jednoj minuti Zaks kaže: „U svakoj se stanici nalazi stvar koja se naziva kratkoročna RNA ili mRNA, koja prenosi kritične informacije iz DNK u našim genima na protein, koji je zapravo stvar od koje smo svi napravljeni. Ovo je kritična informacija koja određuje što će stanica raditi. Dakle, mi o tome razmišljamo kao o operativnom sustavu…. Pa ako biste to zapravo mogli promijeniti…. ako biste mogli uvesti liniju koda ili promijeniti liniju koda, ispostavilo se da to ima duboke implikacije na sve, od gripe do raka.”
Kada se “mijenja” linija koda ili “uvodi” linija koda “(odnosi se na DNA),” kôd “ili DNA se tada mijenjaju, što znači da je pojedinac ili” subjekt “sada promijenio svoj genom u ono što su “znanstvenici ” kodirali. Pojedinac ili subjekt više nije Božja kreacija već kreacija čovjeka, što znači da bi pojedinac ili subjekt mogao biti predmet “patenta”.
Dalje kaže, mRNA će reći stanicama da “kodiraju” protein “virusa”. Ovaj “virusni protein” tijelu je stran. Tijelo pojedinca stvara strani protein koji imunološki sustav napada. Kada tijelo stvara protein koji imunološki sustav tada napada, vaš imunološki sustav napada protein koji stvara vaše tijelo, što znači to se događa u „autoimunskom odgovoru“ ili „autoimunoj bolesti“.
To su nekoliko puta ponovili stručnjaci, liječnici, medicinske sestre i nebrojeni drugi. Kao što čitatelji mogu vidjeti, nitko od nas nije “zviždao Dixie”. Zaks govori o uključivanju ovog sustava; međutim, nema načina da ga isključite. Kada stanice znaju prestati stvarati ovaj “virusni protein”? Stanice ne, dakle, ovo se nastavlja trajno.
U normalnom cjepivu imunološki sustav napada ograničenu količinu “čestica” u adjuvansu stvarajući antitijela ili imunološki odgovor koje tijelo može kasnije prepoznati ako pojedinac dođe u kontakt s istom ili sličnom “česticom”.
- Dr. Lee Merritt On Experimental “Operating System” Injections: We’re Going To See A Lot Of Deaths
- Dr Sherri Tenpenny: FDA Has Broken The Law At Least Twice Concerning COVID Injections (Video)
- Dr. Sherri Tenpenny: How The Depopulation COVID Vaccines Will Start Working In 3-6 Months
- The Vaccine Interview You Really Should Hear To Educate Yourself With Dr. Sherri Tenpenny (Video)
Studiju koju Zaks navodi na 3:12 minute možete pronaći i pročitati na web-uNIH Pub Med Library. U cjelovitom testu studije na ResearchGateu, spominjanje “Luciferaze” događa se na stranici 10. Ključni podaci u ovoj studiji nalaze se na stranici 4 – “Tvorovi imunizirani s 200 mikrograma i izazvani [izloženi gripi H7N9 putem IN (intranazalno)] 49. dana imala je virusno opterećenje ispod razine otkrivanja ”. Ako je virusno opterećenje bilo “ispod razine otkrivanja”, postavljaju se dva pitanja:
1) jesu li tvoji uopće ugovarali H7N9 putem intranazalnog izazivanja; i,
2) ako je virusno opterećenje ispod razine otkrivanja, kako znate da su životinje čak imale virusno opterećenje? To bi dovelo u pitanje učinkovitost injekcije.
Štoviše, studije koje Zaks navodi kako se događaju na ljudima trajale su samo približno 18 mjeseci.
Otprilike u 4:00 minute Zaks započinje raspravu o mRNA cjepivima protiv raka. Odmah nakon toga, Zaks raspravlja o dječjem stanju u kojem nedostaje gen ili “kôd” koji uzrokuje proizvodnju određenog enzima kritičnog za metabolizam gdje je trenutni tretman transplantacija cijelog organa – u ovom slučaju, jetre. Zaks predlaže ubrizgavanje mRNA koja kodira gen koji nedostaje, gen sadržan u DNA na ljudskom genomu, što bi “ispravilo” genetski nedostatak.
Postavite ovo pitanje: što uzrokuje da stanice / tijelo proizvode potrebne enzime / proteine? Zaks na to odgovara izgovaranjem genetskog koda ili DNA. Dakle, mRNA mora izmijeniti genetski kod ili DNK da bi tijelo proizvodilo proteine COV-19, da bi tijelo stvorilo imunološki odgovor.
- Medical Doctors: “You Are Being Played” – “The SARS-C0v2 Virus Does Not Exist”
- The Virus Lies & What They Are Leading Towards With Dr. Andrew Kaufman
- Dr. Lee Merritt: The Vaxx Is Preparing World For Mass Death Event (Video)
- America’s Frontline Doctor Simone Gold Warns Of Risks In “Experimental COVID Vaccine” (Video)
Iz riječi Tal Zaksa iz Moderne, mRNA može promijeniti ljudski genom. Bez obzira na dizajn ili „neželjene posljedice“, ova se tehnologija koristi upravo za to. To naziva “informacijskom terapijom”; iako bi ga neki nazivali “ludom znanošću”. U pokušajima da se “prepiše” genetski kod kako bi se ispravili nedostaci, studije su pokazale da je bilo “kaskadnih kvarova”.
Drugim riječima, promjena jednog “neispravnog gena” u jednom genomu uzrokovala je da drugi geni “propadnu” ili izazovu probleme. I nije samo jedan sljedeći gen postao neispravan, već i mnogi. To je više nego vjerojatno zašto postoji preko 400 štetnih događaja oko eksperimentalne injekcije mRNA.
Dakle, sljedeći put kad netko tvrdi da ta “cjepiva” ne mijenjaju ljudski genom ili DNA, možete uputiti tu osobu Tal Zaksu iz Moderne, Inc. koji tvrdi drugačije.
Pripremila: mr. sc. Arna Šebalj
*
Dr. Madej: če vas genska terapija proti COVID-19 gensko sprememni, sta lahko last proizvajalca: https://www.facebook.com/lidijaw1/videos/1530451883954081
*
SARS–CoV–2 Spike Impairs DNA Damage Repair and Inhibits V(D)J Recombination In Vitro
https://m.planet-lepote.com/konicni-proteini-v-cepivih-za-covid-onemogocajo-popravljanje-okvar-dnk-povzrocajo-raka/
Video o tej študiji: https://www.youtube.com/watch?v=4Unt03UBhbU
*
Tanja Šuligoj: World health summit v Berlinu, otober 2021, Stefan Oelrich, član upravnega odbora družbe Bayer izjavi da je šponta genska terapija; “We are really taking that leap [to drive innovation] – us as a company, Bayer – in cell and gene therapies … ultimately the mRNA vaccines are an example for that cell and gene therapy. I always like to say: if we had surveyed two years ago in the public – ‘would you be willing to take a gene or cell therapy and inject it into your body?’ – we probably would have had a 95% refusal rate,...”.
*
PFIZER CEO: MRNA CHANGING DNA
*
Strokovnjak za patente dr. David Martin: kako so pod pretvezo pandemije uzakonili "gene editing" s CRISPR tehnologijo v covid-19 cepivih (min 37.00): https://forbiddenknowledgetv.net/dr-david-martin-who-they-are-the-names-and-faces-of-the-people-who-are-killing-humanity/?fbclid=IwAR37uwn_4hUzYSmbM1FYocfMXWm-ejQRW_1-mY07A-dTlT-fnTCwV3ESXPM
*
Andrej Kranjc FB 7.12. 2021: Clanek ki pokazuje na moznost dolgorocnih posledic po cepljenju: s protein otezuje popravilo dnk v ljudskih celicah. Dr Tomislav Domazet Loso je navedel en mozen nacin to pa je izgleda se drugi mehanizem po katerem imajo lahko cepiva negativen ucinek na ljudske celice-DNK. Clanek je zasnovan na SARS-CoV 2 virus samo mehanizem je isti kod pri cepivih. Studija iz Svedske.
SARS–CoV–2 Spike Impairs DNA Damage Repair and Inhibits V(D)J Recombination In Vitro Hui Jiang 1,2,* and Ya-Fang Mei 2,*
file:///C:/Users/Uporabnik/Downloads/viruses-13-02056-v2.pdf
*
Dr. Domazet-Lošo: Cepljenje naj se takoj ustavi, mRNA cepiva spreminjajo človeški genom
Hrvaški strokovnjak Dr. Tomislav Domazet-Lošo je v nedavnem videoposnetku izrazil globoko zaskrbljenost nad cepljenjem z mRNA cepivi, prepričan je namreč, da omenjena cepiva zlahka spremenijo človeški genom, medtem ko proizvajalci to dejstvo zanikajo. Navedene trditve so zaskrbljujoče tudi zato, ker gre za priznanega strokovnjaka s področja medicine in genetike. Svoje trditve je podkrepil s strokovnim člankom in videom, ki si ga je v štirih dneh ogledalo 130.000 ljudi
Genski material iz mRNK cepiv se vgradi v človeški genom
Nasprotno s trditvijo proizvajalcev cepiv, Domazet-Lošo, izredni profesor na Medicinski fakulteti Hrvaške katoliške univerze ter višji znanstveni sodelavec na Inštitutu Ruđera Boškovića, pojasni da mRNA nima le možnosti spremeniti genom, temveč to tudi dejansko naredi. Strokovno javnost in odločevalce pa poziva, naj pojasnijo ali vsaj dodatno raziščejo morebitne posledice.
Kaj je biologija retropozicije?
Gre za znanstveno področje, ki je prisotno in se razvija že štiri desetletja, pojasni Domazet-Lošo. Ukvarja se tudi z vgradnja mRNK molekul, kar naj bi bilo že dalj časa poznano znanstveno dejstvo. Kljub temu dognanju, mRNK vakcinologija kot veja znanosti, možnost te vgradnje povsem zanika.
Profesorja je to vodilo v nadaljnje raziskovanje, dopušča namreč možnost, da mRNA vakcinologija, torej veda o cepljenju, pozna mehanizem, ki bi to vgradnjo preprečil.
Profesorja je to vodilo v nadaljnje raziskovanje, dopušča namreč možnost, da mRNA vakcinologija, torej veda o cepljenju, pozna mehanizem, ki bi to vgradnjo preprečil. Kljub raziskavi, takega pojasnila ni našel, še več, v nobeni izmed raziskav na to temo ni zasledil, da bi bili strokovnjaki za razvoj cepiva seznanjeni, da do take spremembe genoma pogosto prihaja.
Po toči zvoniti je prepozno
Raziskovanje področja je pokazalo, da proizvajalci mRNK cepiv proti covidu-19 nikoli niso izvedli testov genotoksičnosti in kancerogenosti, s katerimi to morebitno nevarnost ovrgli. Najmanj, kar bi bilo potrebno storiti, je da strokovnjaki te teste opravijo pred izvedbo globalne cepilne kampanje, je še prepričan Domazet-Lošo.
Ob tem je podrobna analiza sestavin cepiva pokazala, da vsebuje celo sestavine, ki tako vgradnjo še pospešujejo.
Videoposnetek v prispevku celo pokaže prisotnost vgrajenega genskega materiala iz Pfizer-Biontechovega cepiva v človeškem genomu. Ob tem je podrobna analiza sestavin cepiva pokazala, da vsebuje celo sestavine, ki tako vgradnjo še pospešujejo.
Po mnenju predavatelja je raziskovanje teme, predvsem v znanstvenih krogih, onemogočeno, ali pa vsaj nezaželeno.
Ob zapisanem se seveda poraja vprašanje, zakaj takih raziskav v javnosti ni več. Po mnenju predavatelja je raziskovanje teme, predvsem v znanstvenih krogih, onemogočeno, ali pa vsaj nezaželeno.
Je množično cepljenje res varno?
Medtem, ko se nekatere države že odločajo celo za obvezno cepljenje, je odločitev drugod še prepuščena vsakemu posamezniku. Tudi pri nas je veliko prahu dvignilo ugibanje o tem, ali ne bi posameznikom, ki cepiva niso prejeli, bolj intenzivno omejili življenje. Medtem, ko nekateri predstavniki v obveznem cepljenju vidijo edino luč na koncu tunela, drugi strokovnjaki istega področja v tem vidijo grožnjo.
Beljakovinska bodica SARS-CoV-2 poslabša popravljanje poškodb DNK in zavira rekombinacijo V(D)J in vitro
Povzetek
Koronavirus 2 hudega akutnega respiratornega sindroma (SARS-CoV-2) je povzročil pandemijo koronavirusne bolezni 2019 (COVID-19), ki je resno prizadela javno zdravje in svetovno gospodarstvo. Adaptivna imunost ima ključno vlogo v boju proti okužbi s SARS-CoV-2 in neposredno vpliva na klinične izide bolnikov. Klinične študije so pokazale, da imajo bolniki s hudo obliko COVID-19 zapoznele in šibke adaptivne imunske odzive; vendar mehanizem, s katerim SARS-CoV-2 ovira adaptivno imunost, ostaja nejasen. Tukaj z uporabo celične linije in vitro poročamo, da beljakovinska bodica SARS-CoV-2 znatno zavira popravljanje poškodb DNK, ki je potrebno za učinkovito rekombinacijo V(D)J pri adaptivni imunosti. Mehanistično smo ugotovili, da se beljakovinska bodica lokalizira v jedru in zavira popravljanje poškodb DNK z oviranjem ključnih beljakovin za popravljanje DNK BRCA1 in 53BP1 pri rekrutiranju na mesto poškodbe. Naše ugotovitve razkrivajo potencialni molekularni mehanizem, s katerim lahko beljakovinska bodica ovira prilagodljivo (adaptivno) imunost, in poudarjajo potencialne stranske učinke cepiv na osnovi polne dolžine beljakovinske bodice SARS CoV-2.
https://www.ncbi.nlm.nih.gov/labs/pmc/articles/PMC8538446/
***
May 04, 2021
